Епілепсія і панічні атаки. Лікування Епілепсії у дітей. Форми епілепсії дитячого віку
Дебют епілепсії спостерігається переважно в дитячому віці (близько 75% всіх випадків). Епілепсія дитячого віку відрізняється великою кількістю резистентних до лікування форм і поліморфізмом припадків, а також, що особливо важливо, саме в дитинстві за багатьма неясними больовими нападами, пупковими коліками, непритомністю, ацетонеміческімі рвотами можуть ховатися замасковані цими проявами епі
лептіческіе напади органічної природи, на що вказували ще Jackson, Specht і Livingston. І тим не менше епілепсія не повинно бути діагнозом виключення в тих випадках, коли не знайдені ніякі інші діагностичні пояснення. У дітей, які страждають епілептичними припадками, швидко розвиваються функціональні порушення, які потім перетворюються в стійкі зміни характеру, пам'яті, уваги, поведінки і шкільної успішності.
Діагностика та лікування резистентних форм епілепсії у дітей
Інфантильні спазми (ІС) Критерії діагностики ІС:
дебют нападів на 1-му році життя (максимум 4 - 9 міс.);
специфічний характер нападів у вигляді коротких згинальних, розгинальних чи сгибательно-розгинальних скорочень м'язів шиї, тулуба і кінцівок;
висока частота нападів протягом доби, серійність;
затримка психомоторного розвитку різного ступеня вираженості;
специфічний ЕЕГ патерн гіпсарітмія (гіперсінхронізірованний ритм, переважання повільних хвиль високої амплітуди, змішаних з періодами дифузного швидкого ритму, або епізодами уплощения кривої);
резистентність до основних базових антиконвульсанти.
лікування ІС
Базовими препаратами в лікуванні ІС є похідні вальпроєвої кислоти. Середні терапевтичні дози становлять 20 70 мг кг на добу. Проте максимальні дози, які ми застосовуємо, становили 200 мг / кг на добу. Залежно від характеру нападів (спазмів) в якості додаткової до вальпроату терапії призначають такі препарати: бензодіазепіни, ламотриджин. сукцініміди, карбамазепін. Значне поліпшення (купірування нападів на 75 100%) досягалося нами в 78% випадків. При абсолютній резистентності до вказаних препаратів можливе проведення гормональної терапії (АКТГ, синактен-депо, кортикотропін, преднізолон, дексазон) в поєднанні з антиконвульсантами.
Синдром Леннокса Гасто
Критерії діагностики СЛГ (по Lennox - Gastaut - Aicardi):
етіологічна гетерогенність;
дебют нападів у віці 1 7 років;
поліморфізм епілептичних нападів у одного хворого: атипові абсанси, міоклонічні напади (кивки, клювки, здригування), атонически-астатические і тоніко-астатические напади, короткі тонічні судоми, особливо уві сні, клонічні і тоніко-клонічні судоми, рідше парціальні напади);
висока частота нападів протягом доби;
варіабельність нападів по днях (хороші і погані дні);
затримка розумового і мовного розвитку;
ЕЕГ патерн-дифузна повільна пік-хвильова активність білатеральна і синхронна з частотою 1 2,5 Гц, зазвичай з акцентом на лобові і скроневі частки. Сон (повільна фаза) різко провокує патологічну активність типу поліспайк-хвиля з частотою 10 Гц, характерну для тонічних нападів.
лікування СЛГ
Базовими препаратами є похідні вальпроєвої кислоти; середні терапевтичні дози становлять 30 100 міліграм на 1 кг маси тіла. Ефективними комбінаціями в залежності від переважання тих чи інших нападів є: вальпроати + сукцініміди, вальпроати + ламотриджин, вальпроати + карбамазепін.
При наявності генералізованих клонічних (тоніко-клонічних) судом і статусному перебігу нападів, третім препаратом можна призначити похідні барбітурової кислоти. Ефективність лікування становить 70%.
Діагностика та лікування ідіопатичних форм епілепсії
Идиопатические форми епілепсії відносяться в цілому до доброякісним формам. Однак в ряді випадків напади резистентні до базових антиконвульсанти. Підкреслюється недостатня терапевтична ефективність при таких формах, як юнацька абсанс - епілепсія, епілепсія з миоклонические абсансах, епілепсія з міоклонічно астатичними нападами (останні дві форми частіше відносять до криптогенной генералізованої епілепсії).
Дитяча абсанс епілепсія (ДАЕ)
Критерії діагнозу ДАЕ:
дебют в 3 - 8 років;
частіше страждають дівчатка;
типові складні абсанси основний вид нападів;
характерна висока частота нападів: десятки і сотні на добу;
приблизно в 30% випадків можливе приєднання генералізованих судомних нападів;
типовий ЕЕГ патерн генералізована пік-хвильова активність з частотою 3 Гц, що виникає, особливо часто, при гіпервентиляції.
Принципи лікування ДАЕ:
Базові препарати при відсутності генералізованих судомних нападів сукцініміди і вальпроати; при наявності генералізованих судомних нападів виключно вальпроати. Середні терапевтичні дози становлять для сукцінімідов 10 15 мг / кг на добу в 2 прийоми, для вальпроатов 30 50 мг / кг на добу. в 3 4 прийоми. Резервні препарати бензодіазепіни і ламотриджин. У pезістентних випадках застосовуються такі комбінації: вальпроати + сукцініміди; вальпроати + бензодіазепіни; вальпроати ламотриджин. Повна терапевтична ремісія серед обстежених нами хворих була досягнута в 70% випадків і в інших виражене уражень частоти нападів.
Юнацька абсанс епілепсія (ЮАЕ)
Критерії діагнозу ЮАЕ:
дебют нападів, починаючи з 8 років і старше (максимум 9 13 років);
прості типові абсанси (коротші і рідкісні, ніж при ДАЕ) основний вид нападів;
високий ризик приєднання генералізованих судомних нападів до 75%;
на ЕЕГ характерна поява генералізованої пік-хвильової активності з частотою 4 Гц і більше.
Принципи лікування ЮАЕ
Базові препарати виключно похідні вальпроєвої кислоти. Середня терапевтична доза 30 50 мг / кг на добу в 3 4 прийоми. У резистентних випадках, особливо при наявності частих генералізованих судомних нападів, можливі комбінації: вальпроати + барбітурати, вальпроати + ламотриджин. Повна терапевтична ремісія досягається рідше, ніж при ДАЕ, 56% випадків і значне поліпшення 37%. Прогноз погіршується при приєднанні частих генералізованих судомних нападів.
Епілепсія з ізольованими генералізованими судорожними нападами (ГСП).
Критерії діагнозу ДСП:
дебют в дуже широкому віковому інтервалі від 3 до 30 років (в середньому 13 17 років);
проявляється виключно тоніко-клонічними судорожними нападами, зазвичай приуроченим до пробудження або засипанню;
частота нападів невелика, рідко перевищує 1 раз на місяць;
з плином часу можливе приєднання абсансах або міоклонічних нападів з трансформацією в абсанс форми епілепсії або юнацьку миоклоническую епілепсію.
Принципи лікування ДСП
Базовий препарат карбамазепін. Середня дозування становить 15 25 мг / кг на добу в 3 прийоми. Резервні препарати вальпроати, барбітурати, гідантоїни. У резистентних випадках можливі комбінації: карбамазепін + вальпроати; карбамазепін + барбітурати; карбамазепін + гідантоїни: вальпроати + барбітурати; барбітурати + гідантоїни. При приєднанні абсансов або міоклонічних нападів необхідна негайна заміна карбамазепіну на вальпроати. Повна ремісія 70% випадків і значне уражень нападів 27%.
епілепсія- хронічне захворювання головного мозку, проявляю- щееся повторними непровоцірованнимі нападами з порушенням рухових, чутливих, вегетативних, когнітивних, психічних функцій, обумовлених надмірними нейрональними розрядами в сірій речовині кори головного мозку.
Представлене визначення містить два важливих положення: 1) тільки повторні напади є підставою для встановлення діагнозу епілепсії; 2) до епілепсії відносяться спонтанні, непрово- ціруемие напади (виняток становлять рефлекторні форми, наприклад, фотосенсітівная епілепсія). Чи не є на епілепсію фебрильні судоми, а також судоми, що виникають при гострих захворюваннях головного мозку (наприклад, при енцефаліті, субдуральної гематоми, гострому порушенні мозкового кровообігу та ін.).
Сучасні уявлення про захворювання почали складатися тільки з кінця XIX в. Дж. Джексон в 1888 р визначав епілепсію як «... випадкове, раптове і надмірне локальне порушення сірої речовини головного мозку»; описав «ункусние атаки» (нюхові галюцинації при скроневої епілепсії) і «сновідних стану» (напади з порушенням психічних функцій). А Я. Кожевников (1898) розділив всі форми епілепсії на «органічні» (за сучасною термінологією - симптоматичні) і конституціональні (ідіопатичні). Першу спробу класифікації епілептичних нападів зробив англійський невролог В. Говерс в 1903 р Сіндромологіческій підхід в діагностиці епілепсії встановили В. Леннокс в 1961 р, Х. Гасто в 1966 р і Г. Доозе в 1980 р Вагомий внесок у вивчення епілепсії внесли вітчизняні вчені П.М. Сараджішвілі і В.А. Карлов.
В кінці XX в. епілепсія стала виліковним захворюванням. Сучасна класифікація епілептичних синдромів 1989 р констатує, що існує безліч форм епілепсії (синдромів), що мають свої закономірності перебігу і прогноз розвитку в залежності від того, які електричні розряди відбуваються в корі головного мозку, де вони локалізуються, як поширюються і трансформуються і які напади при цьому виникають у хворого. У вивченні епілепсії важливу роль відіграють методи нейровізуалізації (КТ, МРТ з високою роздільною здатністю, ПЕТ, SPECT), цифрова ЕЕГ і відео-ЕЕГ-моніторинг. В даний час приблизно 65% випадків епілепсії повністю виліковні; в 20% випадків це досягається хірургічними методами.
Змінилося і ставлення до хворих, покращилася їх соціальна адаптація. Однак до сих пір багато механізмів патогенезу цього важкого захворювання не вивчені; існує велика кількість атипових форм, значно ускладнюють точну діагностику; як і раніше залишаються некурабельной деякі резистентні форми епілепсії.
Поширеність епілепсії в загальній популяції досягає 05-075%, а в дитячій - 1%. У 75% пацієнтів епілепсія дебютує в дитячому і підлітковому віці, будучи одним з найбільш частих патологічних станів дитячої неврології.
Всі форми епілепсії по етіології підрозділяються на ідіопатичні, симптоматичні і Криптогенні.
для идиопатических форм характерні нормальний інтелект, відсутність вогнищевих симптомів і структурних змін головного мозку у пацієнта, а також спадкова схильність (випадки епілепсії у родичів). Етіологія обумовлена головним чином каналопатій - генетично детермінованої дифузійної нестабільністю мембран нейронів. Ідентифіковано гени трьох основних моногенно успадкованих форм епілепсії: аутосомно-домінантною лобової епілепсії з нічними пароксизмами (локуси 20ql3.2 і 15q24), доброякісних сімейних судом новонароджених (локуси 20ql3.2 і 8q24) і генералізованої епілепсії з фебрильними судомами плюс (локус 19ql3.1 , мутація гена SCN1B; 2q21-q33, мутація гена SCN1A). Інші форми детерміновані кількома генами (полигенное успадкування). До них відносяться юнацька міоклонічна епілепсія, роландична епілепсія, доброякісна парціальна (сімейна) епілепсія дитинства та ін. З практичної точки зору необхідно пам'ятати, що якщо один з батьків хворий ідіопатичною епілепсією, ймовірність народження хворої дитини складе не більше 10%.
симптоматичні форми епілепсії характеризуються обов'язковою наявністю морфологічного субстрату: пухлин, кіст, гліальних рубців, аномалій мозку і аневризм. Їх виявляють за допомогою методів нейровізуалізації.
термін «Криптогенний» ( «Імовірно симптоматичного генезу») визначає ті форми епілепсії, причина яких залишається нез'ясованою навіть при застосуванні всіх сучасних методів об- нання. Наприклад, в разі поєднання епілепсії з гемипарезом або вродженої розумовою відсталістю передбачається симптоматичний характер захворювання, але при КТілі МР-дослідженні зміни в мозку не виявляються.
фокальні напади і форми епілепсії пояснює концепція коркового «епілептогенного вогнища», що грає роль «водія ритму». Виниклий в ньому гіперсинхронних розряд залучає велику кількість нейронів кори, поширюючись на сусідні ділянки головного мозку.
при генералізованих формах епілепсії напади генералізовані з самого початку, що підтверджується даними ЕЕГ (білатерально синхронне поширення на обидві півкулі). Патогенез генералізованих форм епілепсії до теперішнього часу недостатньо ясний. Провідна таламо-кортикальна гіпотеза пояснює виникнення первинної генералізації інтегративної системою, що складається з кори головного мозку і таламуса (таламо-кортикальний і кортико-таламический шляху). Джерело розрядів імовірно знаходиться в корі головного мозку, таламо-кортикальні зв'язку здійснюють синхронізацію генералізованих пік-хвильових розрядів, а ретикулярна формація стовбура (перш за все середнього мозку) модулює рівень «гіперчутливості» кори до розрядів. У поширенні і генералізації епілептичного розряду також беруть участь поясна звивина, орбито-фронтальна кора, амігдала-гиппокампального комплекс, чорна субстанція. При подразненні таламо-кортикальної системи на ЕЕГ може виникати генералізована пік-хвильова активність, а також білатерально синхронні пароксизмальні розряди ритмічних дельта-хвиль.
Первинно генералізована епілепсія виникає за умови аномально високою збудливості таламо-кортикальної системи. Рівень збудливості, ймовірно, детермінується генетично і обумовлений нестабільністю мембран нейронів і неможливістю підтримання нормального градієнта іонів Na, K і Cl.
Класифікація епілептичних нападів була прийнята Міжнародною лігою по боротьбі з епілепсією в 1981 р в Кіото (Японія). Епілептичні напади підрозділяють на: 1) фокальні (вогнищеві, фокальні, локальні, локально зумовлені); 2) генералізовані; 3) не класифікуються (табл. 20).
Фокальні (фокальні, вогнищеві) напади діагностуються в тому випадку, коли на початку пароксизму є чіткі клінічні та електрофізіологічні критерії залучення певних структур головного мозку. Наприклад, при клонических судомах половини обличчя і руки з одного боку (фаціобрахіальние напади) епілептичний вогнище знаходиться в средненіжніх відділах передньої
центральної звивини; при нюхових галюцинаціях - в області гачка скроневої звивини; при Фотопсії - в корі потиличної частки; при «провалах думок» (дісмнестіческіх нападах) - в лобовій долі і т.д. При простих парціальних нападах свідомість не порушено. На ЕЕГ під час нападу відзначається локальний епілептичний розряд, що починається у відповідній області кори великого мозку.
Вогнищевий напад із вторинною генералізацією може починатися як парціальний, але потім переходить в генералізований, залучаючи всі м'язи тулуба і кінцівок, з поширенням епілептиформні активності на ЕЕГ на обидві півкулі.
Складні фокальні напади протікають з порушенням свідомості (під час нападу пацієнт не реагує на звернену мову, не виконує команди, амнезірует напад). ЕЕГ під час складного парціального нападу виявляє одне двосторонній епілептичний розряд, частіше в скроневих або лобових відведеннях (табл. 21).
До генералізований нападів відносять типові та атипові абсанси, клонічні, тонічні, клоніко-тонічні і атонічні напади, а також миоклонии.
Таблиця 20.Міжнародна класифікація епілептичних нападів (Кіото, 1981)

Встановлено, що епілепсія не є єдиним захворюванням з різними нападами, а підрозділяється на окремі форми -
епілептичні синдроми. Вони характеризуються стійкою взаємозв'язком клінічних, електричних і анатомічних критеріїв; розрізняються по реакції на антиепілептичної терапію і за прогнозом (табл. 21).
Таблиця 21.Зміни на ЕЕГ при різних нападах
Таблиця 22.Міжнародна класифікація епілепсій, епілептичних синдромів (Нью-Делі, 1989)
1. Локалізаційно-обумовлені форми епілепсії (фокальні, локальні, фокальні)
1.1. Идиопатические (з возрастзавісімим початком)
Доброякісна епілепсія дитячого віку з центральновісочнимі піками (роландична).
Епілепсія дитячого віку з потиличною пароксизмами.
Первинна епілепсія читання.
1.2. симптоматичні
Хронічна прогресуюча парціальна епілепсія (синдром Кожевникова).
Напади, що характеризуються специфічними способами провокації.
Інші форми епілепсії з відомою етіологією або органічними змінами в мозку.
1.3. Кріптогенние



Слід зазначити, що за минулий після 1989 року час стало очевидно недосконалість класифікації, оскільки в неї не ввійшли деякі форми (наприклад, синдром псевдоленнокса). Крім того, багато симптоматичні форми синдрому Веста і синдрому Леннокса-Гасто не належать до генералізованої епілепсії, оскільки являють собою парциальную епілепсію з феноменом вторинної білатеральної синхронізації. У 2001 р Міжнародна комісія з класифікації та термінології випустила проект нової класифікації епілептичних нападів і епілептичних синдромів (табл. 22). Крім класичного розподілу на фокальні та генералізовані напади, в ньому зазначено, що по відношенню до багатьох доброякісних і самокупірующееся епілептичних синдромів термін «епілепсія» слід заміняти на «напади». Наприклад, не «алкогольна епілепсія», а «напади, пов'язані зі скасуванням алкоголю» і т.д. Описано багато нових форм епілепсії як чітко встановлених, введені нові терміни. Термін «парціальні напади і парціальні епілепсії» замінений на «фокальні напади і фокальні форми епілепсії»; «Криптогенні форми» на «ймовірно симптоматичні форми». У визначенні синдромів рекомендована заміна слова «судоми» на «напади». Поняття «напади» значно ширше поняття «судоми», і далеко не всі напади проявляються саме судомами. Скасовано підрозділ фокальних нападів на прості і складні в залежності від порушення свідомості, так як в більшості випадків оцінка рівня свідомості залишається орієнтовною. Перевагою класифікації є розробка концепції дитячих епілептичних енцефалопатій.
діагностикаепілепсії включає наступний алгоритм:
1. Опис пароксизмального події (можливо виключно за даними анамнезу).
2. Класифікація нападів (анамнез, клініка, ЕЕГ, відеоЕЕГ-моніторинг).
3. Діагностика форми (анамнез, клініка, ЕЕГ, відео-ЕЕГмоніторінг, нейровізуалізація).
4. Встановлення етіології (МРТ, каріотипування, біохімічні дослідження, біопсія м'язів і ін.).
5. Діагностика супутніх захворювань і встановлення ступеня інвалідизації.
Діагноз епілепсії є клініко-електро-анатомічним. У XXI ст. для встановлення точного діагнозу епілепсії недостатньо мати опис нападів, представлене родичами. Необхідно електроенцефалографічне підтвердження (електричний критерій), а також проведення методів нейровізуалізації (анатомічний критерій). Для точного визначення діагнозу і призначення правильної терапії, крім рутинних методик, необхідно проведення тривалого ЕЕГ-відеомоніторингу, нічного ЕЕГ моніторингу, високоразрешающей МРТ в режимі 3D-візуалізації і т.д.
14.1. Идиопатические фокальні форми
Доброякісна парциальная епілепсія дитячого віку з центрально-скроневими піками (роландична епілепсія) [РЕ] - характерізуетсяся короткими фарінгооральнимі і геміфаціальний моторними нападами, що виникають зазвичай при пробудженні і засипанні, а також типовими змінами на ЕЕГ (рис. 14.1). РЕ - найчастіша форма епілепсії в дитячому віці. Показник захворюваності становить 21 на 100 000 дитячого населення.
Захворювання починається у віці від 2 до 14 років (максимум в 7-9 років), частіше хворіють хлопчики. Характерні прості фокальні напади, що виникають в 80% випадків при пробудженні або засипанні. Напад починається з соматосенсорної аури: відчуття поколювання, оніміння з одного боку в області глотки, мови, ясна. Потім пацієнти видають своєрідні горлові звуки типу «булькання», «хрюкання», «полоскання горла»; відзначається гіперса- лівація і анартрия (фарінгооральние напади). Характерні судоми мімічних м'язів: односторонні тонічні, клонічні
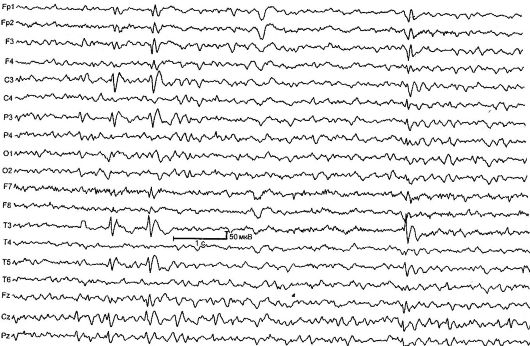
Мал. 14.1.ЕЕГ дитини 4 років з роландической на епілепсію
або тоніко-клонічні судоми м'язів обличчя, губи, а також мови, глотки, гортані (геміфаціальний напади). У 20% хворих судоми поширюються з м'язів обличчя на гомолатеральнимі руку (бра- хіофаціальние напади); приблизно в 8% випадків вони з'являються і в нозі (унілатеральні напади). У міру розвитку захворювання напади можуть змінювати сторонность.
Вдруге-генералізовані судомні напади відзначаються у 25% дітей. Напади при РЕ тривають від кількох секунд до 1-2 хв. Частота їх в середньому - 2-6 разів на рік. З плином часу вони виникають все рідше (навіть без лікування), і у дорослих не спостерігаються.
Зміни на ЕЕГ в межприступном періоді визначаються в 90% випадків, типовий патерн - комплекс гостра-повільна хвиля. Початковий компонент зазвичай складається з трифазного гострої хвилі з подальшою повільною хвилею, що створює подібність з комплексами QRSTна ЕКГ. Ця активність локалізується в центрально-скроневих відведеннях і називається «роландической» або має загальну назву - «доброякісні епілептиформні порушення дитячого віку» (Денді). Для підтвердження діагнозу РЕ важливо проводити
ЕЕГ під час сну - нічний ЕЕГ-моніторинг, так як приблизно у 30% дітей з РЕ роландіческой комплекси виявляються виключно під час сну.
Терапія.З огляду на доброякісний перебіг, годі й назна- чать антиепілептичної терапію. Проте не виключена діагностична помилка, а також можливість трансформації РЕ в синдром псевдоленнокса приблизно в 5% випадків у дітей до 7 років. Рекомендується починати терапію при повторних нападах. Лікування завжди проводять одним препаратом (политерапия неприпустима), починаючи з похідних вальпроєвої кислоти (депакин, конвулекс, КОНВУЛЬСОФІНУ). Вальпроати призначають з поступовим нарощуванням дози до 15- 30 мг / кг на добу (в середньому 600-1500 мг / добу) в 2 прийоми.
При неефективності або непереносимості вальпроатов призначають топирамат (топамакс) в дозі 50-150 мг / сут (3-5 мг / кг). Також застосовуються препарати з групи карбамазепіну (тегретол, финлепсин) в середній добовій дозі 15-20 мг / кг (300-600 мг / добу). В окремих випадках карбамазепін може привести до збільшення індексу Денді на ЕЕГ і почастішання нападів - феномену аггравации. У зв'язку з цим не рекомендується призначати карбамазепін як стартову терапію, а також у всіх випадках у дітей до 7 років. Застосування барбітуратів і гідантоїнів протипоказано!
Необхідний контроль ЕЕГ, в тому числі ЕЕГ-моніторинг сну. Ремісія при РЕ досягається в 100% випадків до 16 років.
Ідіопатична парціальна епілепсія з потиличними пароксизмами (доброякісна потилична епілепсія, ДЗЕ)- характеризується нападами з порушенням зорових функцій, мігренеподобнимі симптомами і наявністю на ЕЕГ паттерна Денді в потиличній області. ДЗЕ становить близько 20% всіх идиопатических парціальних форм епілепсії дитячого віку. Виділено два варіанти ДЗЕ: з ранньої та пізньої маніфестацією захворювання.
Доброякісна потилична епілепсія з раннім дебютом (синдром Панайотопулоса) починається між 1 і 13 роками, з піком маніфестації в 3-6 років. Захворювання проявляється рідкісними важкими нападами з вегетативними порушеннями, тривалою втратою свідомості і тенденцією до статусному течією. Напади виникають уві сні, особливо перед пробудженням; починаються з блювоти, головного болю, збліднення особи, з подальшим поворотом голови і очей в сторону. Напади зазвичай закінчуються геміконвульсівнимі або генералізованими судомами. Виникають «іктальние синкопи», які проявляються тривалої
втратою свідомості і різким падінням м'язового тонусу, тривалістю від 30 хв до 7 ч, в середньому 2 год. Більшість пацієнтів потрапляють в реанімаційне відділення. «Іктальние синкопи» можуть як передувати вторинно генералізований тоніко-клонічним судом, так і виникати ізольовано від них. Незважаючи на важке статусне протягом, частота подібних нападів невелика. У деяких випадках відзначається лише один напад за весь період захворювання. Прогноз - абсолютно сприятливий.
Доброякісна потилична епілепсія з пізнім дебютом (форма Гасто) дебютує з 3 до 15 років, в середньому в 8 років. Характерні прості фокальні сенсорні напади із зоровими порушеннями у вигляді простих зорових галюцинацій (маленьких різнокольорових кругових фігур), які часто виникають в периферичному полі зору і рухаються в протилежну вогнища сторону. Напади тривають від кількох секунд до 1-3 хв. Галюцинації можуть виникати в однойменних половинах полів зору. Часто відзначається версівний компонент - поворот очей і голови контралатерально вогнища при збереженому свідомості. Напади можуть закінчуватися унилатеральной або вторинно генералізованими тоніко-клонічними судомами. У половини пацієнтів після нападу з'являється інтенсивний пульсуючий мігренеподібний головний біль, що супроводжується нудотою і блювотою. Частота нападів зазвичай невелика, хоча в окремих випадках вони можуть бути щотижневими. На ЕЕГ виявляються високоамплітудні комплекси гостра-повільна хвиля, що виникають у 2/3 пацієнтів тільки в потиличних відведеннях. Морфологія комплексів подібна до доброякісними епілептиформними порушеннями дитячого віку. У 1/3 хворих епілептиформна активність може реєструватися і в інших областях (частіше в центрально-скроневих відведеннях).
Терапія.Препаратами першого вибору в лікуванні ДЗЕ є солі вальпроєвої кислоти (депакин, конвулекс, КОНВУЛЬСОФІНУ) в середній добовій дозі 30-40 мг / кг. Препарат призначають в два прийоми з максимальною дозуванням у вечірній час.
При недостатній ефективності можлива монотерапія препаратами карбамазепіну (фінлепсин, тегретол) в середній дозі 15- 20 мг / кг / добу або топіраматом в дозі 75-200 мг / сут (3-6 мг / кг / добу).
При синдромі Панайотопулоса повна ремісія нападів до 9 років настає у 92% пацієнтів. У хворих з формою Гасто реміс- ця спостерігається в 82% випадків до 15 років і в 100% - до 18.
Аутосомно-домінантна лобова епілепсія з нічними нападами
є ідіопатичною формою. Ідентифіковано 2 генних локусу, відповідальних за розвиток даного захворювання: 20q13.2і 15q, але зустрічаються і спорадичні випадки. Вік дебюту варіює від 2 місяців до 52 років, з максимумом на першому десятилітті життя. Напади у 70% пацієнтів починаються з неспецифічної аури: «ознобоподобное тремтіння», головного болю, слухових галюцинацій, головокру- вання, соматосенсорних відчуттів (свербіння в області тулуба), після якої типові напади з гіпермоторние автоматизмами. Вони починаються з судомного дихання, хрюкання, сильного крику за типом завивань. Очі широко відкриті, на обличчі вираз жаху. Пацієнт піднімає голову, сідає в ліжку; з'являються гіпермоторние і дистонічні феномени. Іноді пацієнт (частіше дорослий) соверша- ет хаотичні рухи руками (по типу боксує рухів) і ногами (типу педалювання); встає на карачки і робить розгойдуються руху тазом. Свідомість під час нападів зазвичай не порушено. Характерно виникнення нападів виключно уві сні, вони можуть повторюватися багаторазово протягом ночі у вигляді серії, потім відбувається перерву на кілька днів або тижнів і знову відновлюється серія. Тривалість нападів - від декількох секунд до 1 хв. У виняткових випадках можуть з'явитись вторічногенералізованних пароксизмів.
ЕЕГ неспання неспецифічна. Діагностично значимі дані ЕЕГ-моніторингу нічного сну і відео-ЕЕГ-моніторингу, які виявляють низькоамплітудні епілептиформні активності у вигляді комплексу гостра-повільна хвиля, що виникає регіонально в одному з лобових, лобно-скроневих відведень або біфронтально асинхронно.
Стартове лікування починається з препаратів карбамазепіну, дворазово з максимумом перед нічним сном. Добова доза - 600-1000 мг / добу (15-30 мг / кг / добу). При неефективності призначається топирамат в дозуванні 100-400 мг / сут (3-10 мг / кг / добу), дворазово з максимумом перед нічним сном. Наступний етап лікування - монотерапія вальпроатами. Призначається конвулекс дворазово в дозі
900-1800 мг / добу (20-40 мг / кг / добу).
У рідкісних випадках резистентності може бути застосована политерапия, що складається з комбінації двох базових АЕП (вальпроєвоїкислоти з карбамазепіном або топіраматом). Медикаментозна ремісія досягається в більшості випадків.
14.2. Симптоматичні фокальні форми епілепсії
Симптоматична лобова епілепсія (СЛЕ) - локально обумовлена форма з верифікованим морфологічними порушеннями в межах лобових часток великого мозку. Становить 30-40% серед усіх симптоматичних фокальних форм епілепсій і займає 2-е місце по частоті після скроневої епілепсії (в дитячому віці може випереджати скроневу епілепсію по частоті).
Етіологія включає черепно-мозкові травми, пухлини і кісти лобової частки, фокальні кортикальні дисплазії, глиоз як слід- ствие перинатальної енцефалопатії, судинні аномалії.
В рамках СЛЕ виділяють кілька форми.
Моторна (премоторная, джексоновская) СЛЕ виникає при подразненні передньої центральної звивини. Характерні прості фокальні моторні напади з судомами в контралатеральних вогнища кінцівках. «Джексоновская» марш починається судомами кисті або стопи, з поступовим залученням руки, ноги і м'язів обличчя однойменної боку. Нерідко напад закінчується тимчасовим парезом Тодда.
оперкулярной СЛЕ виникає при подразненні оперкулярной зони лобової частки. Характеризується складними фокальними (діалептіческімі) нападами з оро-аліментарним автоматизмами; можливі іпсилатеральний сіпання лицьової мускулатури, вегетативні феномени.
орбітофронтальная СЛЕ виникає при подразненні орбітальної кори нижньої лобової звивини. Характеризується складними фокальними, вегетативно-вісцеральними нападами, пароксизмами з насильницької вокалізацією, атиповими абсансами.
Дорсолатеральна (префронтальна) СЛЕ виникає із задніх відділів верхньої і нижньої лобової звивини. Виявляється тонічними адверсівних нападами з поворотом очей і голови в бік, протилежну вогнища; можливо також відведення і пріподніма- ня руки, на яку спрямований погляд хворого. Нерідко поява моторної афазії при локалізації вогнища в домінантною гемісфери.
Фронтополярная СЛЕ виникає при локалізації епілептогенного вогнища в області полюса лобових часток. Представлена простими парціальними нападами з порушенням когнітивних функцій (наплив думок, «провал» думок, зміна плину часу) і складними парціальними (діалептіческімі) нападами.
цінгулярной СЛЕспостерігається при подразненні передньої частини поясної звивини. Виявляється складними парціальними нападами з жестовими автоматизмами, ипсилатерально моргательнимі рухами, а також «лимбическими пароксизмами»: виразом страху, почервонінням особи, порушенням емоційної сфери - дисфорией.
СЛЕ, яка виходить із додаткової моторної зони (премоторная СЛЕ), - одна з найбільш частих форм лобової епілепсії, характеризується короткими постуральними асиметричними тонічними нападами (спазмами), що з'являються білатерально в проксимальних відділах кінцівок (наприклад, типу «пози фехтувальника»). Напади переважно нічні, виникають серійно. Також спостерігаються напади із зупинкою мови при ясній свідомості або вокалізацією у вигляді криків, завиваючих звуків. Можливі напади зі стереотипними гіпермоторние автоматизмами: хаотичні рухи руками (по типу боксування), ногами (педалює руху), тазом.
Напади короткі, з нетривалим або неповним виключенням свідомості, мінімальної постіктальном спутанностью, серій- ним ціклолептіческім перебігом і переважним виникненням в нічний час.
Результати неврологічного обстеження залежать від етіології СЛЕ. При великому ураженні лобової частки (наприклад, об'ємному освіту) виявляється гемипарез на стороні, протилежній вогнищу (високі рефлекси, патологічні рефлекси); можлива гемиатаксия. Нерідко формується порушення поведінки по типу «лобової психіки».
ЕЕГ в межприступном періоді малоинформативно або неспецифічно. Переважно тривалий ЕЕГ-моніторинг (і обов'язково під час сну), який виявляє регіональні епілептиформні патерни (гостра-повільна хвиля), продовжене регіональне уповільнення в одному з лобових відведень, феномен вторинної білатеральної синхронізації.
Для виявлення структурного дефекту проводять МРТ.
Стартове лікування починається з топирамата (топамакс) в початковій дозі 12,5-25 мг / сут. Дозу поступово збільшують на 12,5-25 мг 1 раз на тиждень до 50-500 мг / сут (3-10 мг / кг / добу), в 2 прийоми (вранці та ввечері) з інтервалом в 12 ч. Препарат другого вибору - карбамазепін, застосовують в дозі 600-1800 мг / добу (15-35 мг / кг / добу), 2 рази на добу. Карбамазепін і окскарбазепін особливо ефективні при діалептіческіх нападах. При «псевдогенералізованних прісту-
пах »і феномен вторинної білатеральної синхронізації на ЕЕГ карбамазепін протипоказаний, оскільки здатний аггравіровать напади.
Кошти третього вибору - препарати вальпроєвої кислоти (конвулекс, депакин, КОНВУЛЬСОФІНУ) застосовують в дозі 1000-3000 мг / добу (30-60 мг / кг / добу), 2 рази на добу.
При неефективності трьох базових препаратів рекомендована политерапия - комбінація топирамата або вальпроатов з сукці- Німід. Етосуксимід (суксилеп) призначають в дозах 500- 1000 мг / добу (20-40 мг / кг / добу) в 3 прийоми. В інших випадках призначають комбінацію базових АЕП: топирамат + вальпроати, вальпроати + карбамазепін, карбамазепін + топірамат.
Резервні препарати при политерапии - ламотриджин (ламіктал) і леветірацетам (Кеппра). Ламотриджин (3-7 мг / кг / добу) застосовують тільки в комбінації з базовими АЕП. Середні дозування - 100-400 мг / сут в комбінації з топіраматом або карбамазепіном і 100-200 мг / сут з вальпроатами. Леветірацетам ефективний в комбінації з базовими АЕП в дозі 1000-4000 мг / добу (30-60 мг / кг / добу) при фокальних моторних і вторинно генералізованих нападах.
Прогноз захворювання при СЛЕ завжди серйозний, що пов'язане з наявністю структурного дефекту в корі, гемипареза і виражених когнітивних порушень. Медикаментозна ремісія досягається тільки у 20% хворих. В інших випадках вдається істотно знизити частоту нападів. При резистентних нападах застосовується хірургічне лікування. Основний вид оперативного втручання - фокальна кортикальна резекція.
Симптоматична скронева епілепсія (СВЕ) - локально обумовлена форма з відомою етіологією і морфологічними порушеннями в скроневих частках головного мозку (склероз Амона роги, доброякісні вроджені пухлини скроневої частки, фокальні коркові дисплазії, наслідок перинатального ураження). Виділяють дві основні форми СВЕ: лімбічну (синоніми: палеокортікальная, амігдала-гиппокампального) і неокортикальної (синонім: латеральна).
У 75% випадків напади починаються з аури.Слід чітко визначити поняття аури і відмежувати її від провісників епі- лептіческіе нападу. Під аурою (від грец. - подих) слід розуміти клінічні феномени, які виникають самі по собі
або перед вторинно генералізованим або парціальним приступом. Аура обумовлена локальним епілептичних розрядом в певній ділянці кори великого мозку і по суті є простим парціальним приступом. Характер аури вказує на локалізацію вогнища. Виділяють наступні види аури: соматосенсорную, зорову, нюхову, смакову, слухову, запаморочення, психічну, вегетативну, черевну (абдоминальную). передвісникивиникають за багато хвилини, години або дні до епілептичного нападу, про- є зазвичай психічними або вегетативними симптомами, які не супроводжуються локальними кортикальними розрядами.
Амігдала-гиппокампального (палеокортікальная, лімбічна) - найбільш часта форма, становить близько 65% серед усіх випадків СВЕ. В основі захворювання частіше лежить склероз (глиоз) медіобазальних відділів скроневої частки внаслідок перинатального ураження або атипових фебрильних судом. Захворювання зазвичай починається з тривалих, нерідко геміклоніческіх, фебрильних судом у віці до 3 років. Далі слідує період уявного благополуччя - при- ступи відсутні аж до препубертатного періоду. Найбільш типові (70% випадків) складні фокальні напади з виключенням свідомості (діалептіческіе) або автоматизмами (аутомоторние). При діалептіческіх нападах хворий раптово припиняє рухову активність, застигає з широко розкритими очима, погляд висловлює здивування або переляк ( «staring gaze»).
Для СВЕ характерні автоматизми у вигляді жестів (потирання рук, пальців, стискання кисті, перебирання одягу) і оро-аліментарних дій (щебет, ковтання, облизування). Автоматизми в кисті спостерігаються на стороні вогнища, а дистоническая установка пальців кисті - на протилежній. Тривалість аутомоторних нападів від 30 сек до 3 хв, вони швидко частішають і стають резистентними до терапії.
Нерідко напади супроводжуються порушенням вегетативних функцій. Особливо характерні епігастральні пароксизми при ясній свідомості. Пацієнт відчуває біль, розпирання, дискомфорт в області пупка; можливо відходження газів. Це «висхідний епілептичний відчуття» піднімається з живота вгору до горла, супроводжується відчуттям стиснення шиї, після чого можливо вимикання свідомості.
Також характерні прості фокальні напади з порушенням психічних функцій: сновідних стану Джексона ( «dreamy states»), які проявляються раптовими своєрідними відчуттями
«Снів наяву»; відчуття «вже баченого» або «ніколи не баченого»; виникнення дереалізації (відчуття нереальності навколишнього) або деперсоналізації (порушення сприйняття власної особистості). При залученні мигдалеподібного комплексу з'являються короткі напади невмотивованого страху, дисфорії, агресії.
Латеральний (неокортикальної) СВЕ виникає при ураженні Верхньолатеральна відділів скроневої частки. Можливі такі види нападів: слухові галюцинації (пароксизмальні відчуття шуму, музики, голосів); зорові галюцинації (пароксизмальное поява складних яскравих панорамних зорових образів, нерідко з елементами спогади минулих подій); напади несистемного запаморочення, часто в поєднанні з вегетативними проявами (блідість шкіри, гіпергідрозом, тахікардією); пароксизмальна сенсорна афазія при локалізації епілептогенного вогнища в домінантному півкулі; «Скроневі синкопи» з виключенням свідомості, обмяканіем і повільним падінням без судом.
При неврологічному огляді нерідко виявляються пірамідні симптоми контралатерально вогнища: порушення функції VII і XII черепних нервів, асиметрія м'язового тонусу, анизорефлексия, патологічні рефлекси. У дорослих пацієнтів при тривалому перебігу захворювання розвиваються особистісні і когнітивні порушення, що позначаються терміном «глішроідія»: в'язкість, тугоподвижность, інертність мислення, складності перемикання, «застрявання» на дрібницях, стійкість афекту; зниження пам'яті та уваги.
ЕЕГ в межприступном періоді в 50% випадків - без патологічних змін. Пік-хвильова активність в скроневих частках регі- стріруется не більше ніж у 20% пацієнтів.
На МРТ в коронарної проекції можуть виявлятися склероз гіпокампа, розширення нижнього рогу бічного шлуночка, зменшення в обсязі ураженої скроневої частки, в ряді випадків - фокальна кіркова дисплазія.
Лікування починають з препаратів карбамазепіну (фінлепсин ретард, тегретол СR), в дозі 600-1800 мг / добу (15-35 мг / кг / добу) в 2 прийоми з 12-годинним інтервалом або в 3 прийоми з 8-годинним інтервалом. Окскарбазепін (трілептал) призначають в дозі 600 2400 мг / добу (20-40 мг / кг / добу). Препарат другого вибору - топирамат, призначають, поступово збільшуючи дозу до 100-400 мг / добу (4-8 мг / кг / добу), 2 рази на день.
Кошти третього вибору - препарати вальпроєвої кислоти застосовують в дозі 1000-3000 мг / добу (30-70 мг / кг / добу) в 2 або 3 прийоми з рівними інтервалами часу.
При неефективності трьох базових препаратів рекомендована политерапия: комбінації карбамазепіну (або окскарбазепіну) з вальпроатами, топираматом; вальпроатов з топіраматом. Резервні препарати при политерапии - ламотриджин (3-7 мг / кг / сут, тільки в комбінації з базовими АЕП) і леветірацетам.
П рогноз.Медикаментозна ремісія досягається лише у 1/3 хворих. В інших пацієнтів в більшості випадків вдається істотно знизити частоту нападів. У медикаментозно резистентних випадках застосовують хірургічне лікування, зокрема селективну амігдала-гіппокампотомію.
Симптоматична потилична епілепсія (СЗЕ) характеризується наявністю епілептогенного вогнища і морфологічними змінами в потиличній області. Етіологічними факторами є фокальні коркові дисплазії, наслідок перинатальних уражень, окціпітальной кальцифікати з целіакію, судинні аномалії (синдром Штурге-Вебера), MELAS, прогресуюча міоклонус-епілепсія з тільцями Лафора, пухлини, ГПМК в басейні задньої мозкової артерії.
Вік початку СЗЕ вариабелен. Констатують такі види нападів: прості фокальні сенсорні із зоровими рас розладами (макро-, мікропсіі, елементарні зорові галюцинації), з окоруховими порушеннями (адверсія голови і очей в протилежну вогнища сторону, форсоване пароксизмальное моргання, ністагм); вегетативно-вісцеральні (нудота, блювота, головний біль); вторинно генералізовані судомні. Нерідко в структурі нападу (або як постпріступном симптомів випадання) спостерігаються сліпота і гомонимная квадрантная гемианопсия. Характерна постпріступном мігренеподібний головний біль.
При неврологічному обстеженні в окремих випадках визначаються косоокість, амбліопія, звуження полів зору або гемианопсия. ЕЕГ-дослідження в міжнападу у 30% хворих СЗЕ не виявляється патологічних змін. Найчастіше визначаються регіональне уповільнення або пік-хвильова епілептиформна активність в одному з потиличних відведень або біокціпітально з амплітудним переважанням на стороні вогнища.
Нейровізуалізація виявляє потиличні кортикальні дисплазії, локальний глиоз внаслідок перенесеної перинатальної енцефалопатії (улегірія), кальцифікати, судинні аномалії.
лікуванняпочинають з препаратів карбамазепіну в дозі 600- 1800 мг / добу (15-35 мг / кг / добу), в 2 прийоми з 12-годинним інтервалом. Карбамазепін в високих дозах особливо ефективний при ізольованих зорових Аурах і фокальних нападах з порушенням вегетативних функцій. Багато авторів рекомендують починати лікування СЗЕ з окскарбазепіну в дозі 600-2400 мг / добу (20-40 мг / Ксут).
Препарат другого вибору - топирамат призначають в дозі 100 400 мг / добу (5-8 мг / кг / добу) 2 рази на день. При вторинної білате- ральной синхронізації на ЕЕГ топамакс може бути стартовим препаратом.
Препарат третього вибору - вальпроєва кислота. Середні дозування - 1000-2000 мг / добу (30-60 мг / кг / добу), при необхідності - вище, в 2 або 3 прийоми.
У резистентних випадках застосовується политерапия. Особливо ефективні комбінації карбамазепіну (або окскарбазепіну) з вальпроатами, вальпроатов з топіраматом, рідше - карбамазепіну з топіраматом. При додаванні другого препарату дозування першого, як правило, не зменшується. Резервні препарати при политерапии - ламотриджин і леветірацетам.
прогноззалежить від характеру структурного дефекту мозку і шляхів поширення збудження в корі. У 40-50% хворих може бути досягнута стійка медикаментозна ремісія. У резистентних випадках СЗЕ при відсутності ефекту від застосування АЕП єдиним методом реальної допомоги пацієнтам є нейрохірургічне втручання - кортикальна резекція.
Епілепсія Кожевникова і енцефаліт Расмуссена (ЕК) - поліетіо- логічне захворювання, що виявляється поєднанням миоклонических, фокальних моторних, вторинно генералізованих нападів з вогнищевими неврологічними симптомами.
Захворювання вперше описав російський невролог професор Олексій Якович Кожевников під назвою «epilepsia corticalis sive partialis continua». 21 січня 1894 на засіданні створеного ним Московського товариства неврологів і психіатрів він виступив з доповіддю на тему «Про особливий вид кортикальной епілепсії». Доповідь було засновано на вивченні 4 випадків кортикальной епілепсії, які спостерігаються автором в клініці нервових хвороб Москви, і представляв собою
оригінальне опис захворювання, до того часу ще не відомого. Клінічна картина хвороби у всіх 4 пацієнтів була надзвичайно схожа: «... поєднання генералізованих епілептичних при- ступити з постійними клонічними судомами в суворо визначених частинах тіла. З цих постійних судом розвивалися: 1) типові джексоновские припадки в одній половині тіла і 2) вищезазначені загальні припадки, що розвивалися також по джексоновская типу ». Інша назва цього захворювання було запропоновано присутнім на доповіді професором Н.Ф. Філатовим - «кожевніковская епілепсія». У 40-ті роки минулого століття була доведена взаємозв'язок ЕК з весняно-літнім кліщовим енцефалітом (російський енцефаліт).
У 1958 р Т. Расмуссен, Ж. Обжевскі описали клініку хронічного осередкового енцефаліту, одним з кардинальних симптомів якого була ЕК. Пізніше дане захворювання було названо енцефалітом Расмуссена, або синдромом Расмуссена (СР). До теперішнього часу залишається загадкою, при якому захворюванні А.Я. Кожевников описав симптомокомплекс ЕК - при російській енцефаліт або енцефаліт Расмуссена. На нашу думку, А.Я. Кожевников, який практикував в Москві, описав свою форму епілепсії саме при хронічному очаговом енцефаліт, так як ні в одній з поданих ним історій хвороби немає вказівок на перенесений пацієнтами гострий енцефаліт.
Крім кліщового енцефаліту, ЕК викликають туберкульозний менінгоенцефаліт, нейросифилис, черепно-мозкова травма, опу- холи головного мозку, фокальні кортикальні дисплазії, спадкові хвороби обміну.
Хронічний вогнищевий енцефаліт [енцефаліт Расмуссена, синдром Расмуссена (СР)]. СР є тяжке захворювання головного мозку - хронічний прогресуючий осередковий енцефаліт. Захворювання характеризується тріадою клінічний симптомокомплекс: епілептичними нападами (по типу епілепсії Кожевнікова), руховими порушеннями (центральний геміпарез) і розладом вищих психічних функцій. Етіологія невідома, імовірно захворювання відносять до повільним нейроінфекції вірусної етіології, але вірус не ідентифікований.
Дебют в дитячому віці - від 1 року до 14 років, з піком в 5-6 років з епілептичних нападів (фокальних моторних або вторічногенералізованних, рідше - діалептіческіх); в 20% випадків - з епілеп-
тичного статусу. Нерідко відзначається соматосенсорная аура (печіння, поколювання, оніміння). Уже на початкових етапах захворювання розвивається тимчасовий постіктальном монопарез (або геміпарез) - парез Тодда. Зазвичай через кілька місяців після появи перших фокальних нападів до них приєднуються тривалі (до декількох днів), а потім постійні, локалізовані в одній половині тулуба і кінцівок міоклонічні пароксизми, які можуть трансформуватися в генералізовані судоми. Зазначений симптомокомплекс є епілепсію Кожевникова. З плином часу епілептичний миоклонус поширюється на всі кінцівки, лицьову мускулатуру, м'язи передньої черевної стінки і стає постійним, зникаючи і уві сні. Розвивається стійкий гемипарез. Приєднуються порушення чутливості по проводниковому типу і випадання полів зору. Наростають когнітивні порушення, дизартрія. У 25% випадків можливе ожиріння, преждев- ремінне статевий розвиток.
На ЕЕГ в розгорнутій стадії захворювання в 100% випадків спостерігається прогресуюче уповільнення основний активності фону, продовжене регіональне уповільнення (в лобно-скроневих відведеннях); продовжена пік-хвильова активність. У міру прогресування епілептиформна активність виникає дифузно.
Нейровізуалізація має вирішальне значення в діагностиці. При МРТ головного мозку в динаміці відзначається наростання геміа- трофей. Атрофія зазвичай починається з тім'яно-скроневої області у вигляді локального розширення сильвиевой щілини і з плином часу поширюється «подібно масляного плямі по листу пергаментного паперу », захоплюючи« здорове »півкуля.
ЕК відноситься до резистентним епілептичним синдромам. Стартова терапія - вальпроати (депакин, конвулекс, КОНВУЛЬСОФІНУ) в високих дозах: до 50-100 мг / кг / сут. Далі рекомендується комбінація вальпроатов з леветірацетама або топіраматом. Показана ефективність леветірацетама при фокальних моторних, вторинно генералізованих і міоклонічних нападах в рамках ЕК, його дозування - 30-70 мг / кг / сут. Дозування топирамата становить близько 10 мг / кг / сут. В розгорнутій стадії захворювання можливе застосування барбітуратів (фенобарбітал 5-8 мг / кг / добу). Додавання етосуксимід (до 30 мг / кг / добу) до базових АЕП в окремих випадках може бути ефективно при резистентних миоклонических нападах.
Бензодіазепіни (клобазам 1 мг / кг / добу або клоназепам 0,5 4,0 мг / добу) застосовують у пацієнтів з серійними нападами і статусним плином. Призначення карбамазепіну в якості монотерапії НЕ реко- мендовано зважаючи на можливу аггравации миоклонических нападів.
У лікуванні самого енцефаліту застосовуються різні медикаментозні препарати: антивірусні (зидовудин, ацикловір, ганцикловір); гормональні (метилпреднізолон внутрішньовенно 400 мг / м 2 поверхні тіла протягом 3 днів; преднізолон, дексаметазон); імуноглобуліни (октагам, IVIC 400 мг / кг / добу внутрішньовенно протягом 3 днів); цитостатики (азатіоприн, циклофосфан), плазмаферез. Однак дане лікування може лише уповільнити прогресування захворювання.
Ефективно нейрохірургічне втручання - функціональна гемісферотомія, яка повинна бути виконана як можна раніше. Частота стійкої ремісії після операції становить 23-52%. без оперативного лікування СР прогресує і закінчується летально протягом 2-15 років (в середньому через 3 роки) з моменту дебюту. Описано окремі випадки спонтанної стабілізації захворювання.
14.3. Идиопатические генералізовані форми епілепсії
Доброякісна міоклонічна епілепсія дитинства дебюті- рует у віці від 4 місяців до 3 років. Характерні виключно міоклонічні напади у вигляді активного міоклонусу в м'язах шиї і проксимальних відділах верхніх кінцівок: короткі кивки з легким нахилом тулуба вперед, миттєвим підведення плечей і розведенням ліктів в сторони. Зазвичай напади серійні, дедалі частіші після пробудження. Свідомість не порушено. Значно рідше спостерігаються миоклонические напади в нижніх кінцівках - миттєве згинання ніг з легким присіданням і навіть можливим раптовим падінням на сідниці.
У неврологічному статусі виявляються м'язова гіпотонія і атаксія. Психомоторне розвиток не страждає. На ЕЕГ основна активність не змінена; епілептиформна активність реєструється тільки в момент нападу. Характерні короткі розряди генералізованої полипик-хвильової активності, що виникає синхронно з миоклоническими нападами. Для реєстрації коротких миоклонических нападів незамінний метод відео-ЕЕГ-моніторингу. Зміни при нейровізуалізації відсутні.
Стартове лікуванняздійснюється препаратами вальпроєвої кислоти. Призначають конвулекс або депакин в сиропі або краплях (після 1-2 років - таблетовані препарати) в дозуванні 300-1500 мг / добу (15-50 мг / кг / добу). У більшості випадків настає ремісія. При неефективності застосовують політерапію; при цьому вальпроати завжди залишаються базовими АЕП. Призначають комбінацію вальпроатов з сукцініміди (етосуксимід в дозі 250-750 мг / сут, 15-25 мг / кг / сут, в 2-3 прийоми). Можливі комбінації вальпроатов з топіраматом в дозі 25-100 мг / сут (3-5 мг / кг / добу) в 2 прийоми; вальпроатов з бензодіазепінами, наприклад, клобазам (фрізіум) в дозі 5-20 мг / сут (0,5-1,0 мг / кг / добу) в 2 прийоми. Призначення карбамазепіну і ламо- тріджіна обмежена через можливість аггравации миоклонических нападів.
прогнозсприятливий. Психічне розвиток не страждає, і медикаментозна ремісія настає практично в 100% випадків. Тривалість терапії - 3 роки, рецидиви трапляються рідко.
Епілепсія з миоклонические-астатичними нападами (синдром Доозе) дебютує у віковому інтервалі від 1 до 5 років, частіше з генералізованих судомних нападів, що виникають в будь-який час доби. У 11% випадків в анамнезі відзначаються фебрильні судоми. Типові міоклонічні та міоклонічно-астатические напади приєднуються зазвичай тільки після 3 років. Напади характеризуються короткими, блискавичними, зазвичай асинхронними і аритмічний посмикуваннями в ногах і руках, частіше в проксимальних відділах. Характерно поява миоклонических «кивків», поєднаних з легкої пропульсіі тулуба і при- підніманням плечей ( «активні кивки»). Частота миоклонических нападів може бути дуже високою; нерідко напади виникають багаторазово протягом однієї хвилини або навіть постійно, особливо після пробудження (епілептичний статус). При міоклонічних нападах в нижніх кінцівках виникають каскадні присідання з можливим раптовим падінням на коліна або сідниці (міоклонічно-астатические напади); при цьому свідомість збережена. Абсанси спостерігаються у 60-90% хворих. Переважають короткі типові прості абсанси, а також абсанси з міоклоніческім компонентом. Частота абсансов висока, з максимумом в ранкові години.
У неврологічному статусі відзначаються односторонні пірамідні симптоми, координаторні порушення; в половині випадків - груба
затримка психомовного розвитку. На ЕЕГ виявляються короткі генералізовані і регіональні розряди пік-і полипик-хвильової активності. Зміни при нейровізуалізації, як правило, відсутні; в деяких випадках констатується помірна субатрофия кори.
Стартове лікуванняздійснюється препаратами вальпроєвої кислоти в дозі 600-1750 мг / добу (20-100 мг / кг / добу). Препаратом другого вибору є топирамат в 2 прийоми в дозуваннях 50-200 мг / сут (3-7 мг / кг / добу). При неефективності застосовується политерапия; при цьому спочатку вальпроати, а потім топирамат залишаються базовими АЕП. Застосовують комбінацію вальпроатов з сукцініміди, вальпроатов з топіраматом, вальпроатов з бензодіазепінами. В окремих резистентних випадках можливе призначення трьох АЕП: вальпроатов, топирамата і сукцінімідов (або бензодіазепінів). Застосування карбамазепіну протипоказано через можливість аггравации миоклонических нападів.
Прогноз.У більшості дітей вдається купірувати напади. Приблизно у 1/3 пацієнтів епілептичні напади зберігаються, приєднуються тонічні напади і атипові абсанси, поглиблюється когнітивний дефект.
Абсансние форми епілепсії. Найбільш частими і добре вивченими абсансная формами є дитяча та юнацька абсансепілепсіі. Вони проявляються типовими абсансами - короткими первинно-генералізованими нападами з виключенням свідомості, завмиранням, мінімальними руховими феноменами і наявністю на ЕЕГ симетричною білатерально синхронної пікволновой активності з частотою 3 і більше комплексів в секунду (рис. 14.2). Розрізняють прості (завмирання без рухового компонента) і складні (з мінімальними руховими феноменами) абсанси. До складних належать абсанси з тонічним (відхилення голови назад, заклад очей вгору), міоклоніческім (здригання, сіпання повік, брів, крил носа, плечей), атоническим (падіння голови на груди, нахили тулуба), вегетативним (зміна кольору шкірних покривів, Мимовільне сечовипускання), а також з асиметричними проявами (наприклад, з легким поворотом голови). Тривалість абсансних нападів становить від 2 до 30 с, частота - до 100 і більше на добу.
Дитяча абсанс-епілепсія (пікнолепсія) - найбільш часта форма абсансной епілепсії. Картіровани мутантні гени ГАМК-рецептора в
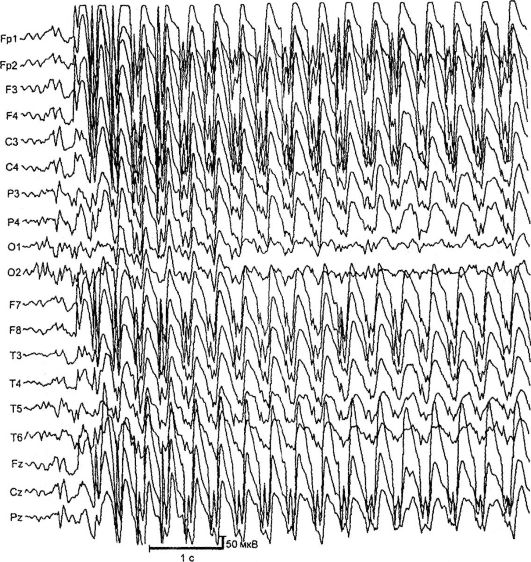
Мал. 14.2.ЕЕГ під час нападу (абсанс)
декількох локусах хромосом: 6р, 8q24, 15q24. Захворювання дебютує у віці 3-9 років з типових абсансов. У рідкісних випадках захворювання починається з генералізованих судомних нападів з подальшим приєднанням абсансов. Частіше хворіють дівчатка. Характерний тип нападів - абсанси з тонічним компонентом: легке закидання голови і заклад очних яблук. Напади провокуються гіпервентиляцією, рідше - усним рахунком. При неадекватному лікуванні приблизно у 30% хворих приєднуються ГСП. На ЕЕГ при проведенні гіпервентиляції з'являються продовжені генералізовані розряди пік-хвильової активності з частотою 3 Гц. МРТ змін не виявляє.
Антіабсансной активністю володіють: вальпроати, сукцініміди, бензодіазепіни, ламотриджин, топірамат. застосування препаратів
карбамазепина протипоказано, оскільки вони провокують почастішання нападів. Стартове лікування здійснюється препаратами вальпроєвої кислоти 2 рази на добу, в дозуванні 600-1800 мг / добу (30-50 мг / кг / добу). У більшості пацієнтів напади повністю купіруються при монотерапії вальпроатами. Препарати другого вибору - сукцініміди. Сукцініміди застосовуються в якості монотерапії при наявності у хворого ізольованих абсансов, дозування етосуксимід - 500-1000 мг / добу (15-30 мг / кг / добу) в 3 прийоми.
У рідкісних резистентних випадках застосовують політерапію: вальпроати + сукцініміди, вальпроати і ламотриджин. Повна тера- певтіческая ремісія досягається в 90-97% випадків, зазвичай при монотерапії. Скасування препаратів починається через 3 роки після припинення нападів.
Юнацька абсанс-епілепсія (ЮАЕ) - форма ідіопатичної генералізованої форми епілепсії, що характеризується типовими абсансами, дебютує в пубертатному періоді з високою ймовірністю приєднання ГСП і ЕЕГ-змінами у вигляді коротких розрядів генералізованої швидкої пік-хвильової активності. Етіологія - мутація гена нікотинового ацетилхолінового рецептора, пов'язаного з хромосомами 5, 8, 18 і 21. Захворювання починається у віці 9-21 року (максимум - в пубертатний період). У 40% випадків епілепсія дебютує з ДСП, в інших - з абсансов. Характерні прості абсанси, меншої тривалості і частоти, ніж при дитячій формі. В окремих випадках виявляють дуже короткі (до 3 с) абсанси з міоклоніческім компонентом: завмирання, легке заклад очних яблук вгору і швидке посмикування повік. У 75% хворих спостерігається поєднання абсансов з ДСП. Судомні напади зазвичай виникають в ранкові години, після пробудження пацієнтів. Частота нападів невелика - 1-4 рази на рік.
ЕЕГ характеризується нормальною основний активністю, на тлі якої виявляються короткі розряди генералізованої швидкої (4 Гц) пік-хвильової активності. Велике діагностичне значення має поява епілептиформні активності при депривації сну, ритмічної фотостимуляції і закриванні очей. При ЮАЕ фотосенсітівная становить 20,5%, а при ДАЕ - 10%. Проба з гіпервентиляцією при ЮАЕ малоинформативна.
Стартова терапія здійснюється препаратами вальпроєвої кислоти в дозі 900-2000 мг / добу (30-40 мг / кг / добу) в 2 прийоми. при
відсутності ефекту від монотерапії переходять на комбіновану терапію (вальпроати + топирамат, вальпроати + сукцинімід).
Повна терапевтична ремісія досягається в середньому у 70% хворих. Скасування терапії здійснюється поступово, не менше ніж через 4 роки повної відсутності нападів.
Епілепсія з ізольованими генералізованими судорожними нападами (Епілепсія з генералізованими судорожними нападами пробудження) (ЕГСП) - форма ідіопатичної генералізованої епілепсії, при якій єдиним типом нападів є первинно-генералізовані тоніко-клонічні судомні пароксизми без аури і чіткого фокусу на ЕЕГ. Форма детермінована генами CLCN2 на хромосомі 3q26 і геном CACNB4 на хромосомі 2q22-23.
Дебют захворювання в широкому віковому діапазоні - від 10 до 30 років (максимум - в пубертатному періоді). Генералізовані тоніко-клонічні напади відбуваються без аури, приурочені до періоду пробудження або засипання. Провокуються депривація сну (зменшення загальної тривалості сну, пізній відхід до сну, пробудження в незвично ранній час). Тривалість ГСП - від 30 с до 10 хв, частота їх невелика. У більшості пацієнтів спостерігається не більше 2-5 нападів на рік.
ЕЕГ в межприступном періоді у 50% хворих нормальна. Рекомендується проведення ЕЕГ після депривації сну і нічний відео-ЕЕГ-моніторинг. У міжнападу спостерігаються короткі генералізовані пік-хвильові розряди. Тоническая фаза ГСП характеризується появою на ЕЕГ дифузного, наростаючого за амплітудою швидкого ритму частотою 20-40 Гц, поступово сповільнюється до 10 Гц. Під час клонической фази даний ритм поступово заміщається генералізованої полипик-хвильової активністю. У фазі постпріступном релаксації домінуючою є дифузна дельта-активність; регіональні феномени відсутні.
При ЕГСП відзначається досить висока ефективність всіх основних груп АЕП: барбітуратів, гідантоїнів, карбамазепі- на, окскарбазепіну, вальпроатов, топирамата, леветірацетама. Фенобарбітал і дифенін через виражених побічних ефектів застосовуються в останню чергу при відсутності ефекту від базових АЕП. Базовими препаратами при епілепсії з ДСП є топирамат, вальпроати і група карбамазепіну.
Лікування починають з топирамата в дозі 100-400 мг / добу (4-10 мг / кг / добу) в 2 прийоми. Препарат другого вибору - вальпроєва кислота в дозі 1000-2000 мг / добу (30-50 мг / кг / добу) в 2 прийоми. Препарат третього вибору - карбамазепін або окскарбазепін (трілептал).
В окремих резистентних випадках можлива монотерапія барбітуратами або гидантоинами, що ефективно, але нерідко призводить до розвитку виражених побічних ефектів і зниження якості життя пацієнтів. У рідкісних резистентних випадках доводиться вдаватися до политерапии. Оптимальна комбінація: топирамат + вальпроати; при цьому дози препаратів залишаються незмінними.
Ремісія досягається у 90% хворих. Відсутність ефекту часто пов'язано з неправильною діагностикою. При неадекватному лікуванні можливе приєднання абсансах або міоклонусу з трансформацією в ЮАЕ і ЮМЕ.
Юнацька міоклонічна епілепсія (ЮМЕ - синдром Янца) - форма ідіопатичної генералізованої епілепсії, що характеризується дебютом в підлітковому віці і наявністю масивних миоклонических нападів, що виникають переважно в період після пробудження пацієнтів.
ЮМЕ - гетерогенне захворювання, пов'язане з мутацією кількох генів, що включають GABRA1-ген(OMIM 137160) на хромосомі 5q34-q35, CACNB4-ген(OMIM 601949) на хромосомі 2q22-q23 і мутацію CLCN2-гена (OMIM 600570) на хромосомі 3q26. Ризик возникно- вения епілепсії у дітей в сім'ї, де один з батьків хворий ЮМЕ, становить близько 8%. Генералізована пік-хвильова активність на ЕЕГ констатується у 18% клінічно здорових родичів пробанда, що страждає ЮМЕ.
Захворювання починається у віці від 7 до 21 років з максимумом у віковому інтервалі 11-15 років. Основний вид нападів - міоклонічні пароксизми, що характеризуються блискавичними посмикуваннями різних груп м'язів. Вони частіше двосторонні, симетричні, поодинокі або множинні, мінливі за амплітудою; нерідко з'являються у вигляді серії залпів. Локалізуються головним чином в плечовому поясі і руках, переважно в раз- гібательних групах м'язів. Свідомість під час міоклонічних нападів збережено. У 30% пацієнтів миоклонические напади захоплюють м'язи ніг, при цьому хворий відчуває раптовий удар під коліна і злегка присідає або падає (міоклоніческі- астатические напади). Міоклонічні напади виникають або
частішають в перші хвилини і години після пробудження. Зниження рівня неспання, сонливість, позіхання, прикривання очей підвищують ймовірність появи нападів в ранкові години.
У 90% випадків міоклонічні напади поєднуються з ГСП пробудження - даний вид нападу називається клонико-тоніко- клонічним. У 40% пацієнтів приєднуються короткі абсанси.
Провокуючими факторами є депривація сну і раптове насильницьке пробудження. У деяких пацієнтів міо клонічні напади виникають виключно при недосипанні. Приблизно у 1/3 хворих ЮМЕ (частіше жіночої статі) напади є фотосенсітівная: провокуються переглядом телепередач, комп'ютерними іграми, миготінням світла на дискотеках. Основний ЕЕГ-патерн - короткі розряди генералізованої швидкої полипик-хвильової активності, що виявляються у 80-95% хворих в міжнападу. Найбільш типова генералізована швидка (4 Гц і вище) полипик-хвильова активність. ЕЕГ при ЮМЕ необхідно проводити рано вранці після ночі з депривації сну.
Диференціальний діагноз ЮМЕ проводять з тиками, хореей, а також з різними формами прогресуючих епілепсій з міоклонусом. Поряд з медикаментозною терапією необхідно строго дотримуватися режиму сну і неспання; уникати факторів фотостимуляції в побуті.
Стартове лікування - препарати вальпроєвої кислоти в дозуванні 1000-2500 мг / добу (30-50 мг / кг / добу). Для того щоб уникнути побічних ефектів у дівчат (порушення менструального циклу, ожиріння, гірсутизм, полікістоз яєчників, зниження фертильності), лікування можна починати з топирамата або леветірацетама у вигляді монотерапії. Топірамат призначають в дозі 200-400 мг / добу (5-10 мг / кг / добу) в 2 прийоми. Леветірацетам призначається в дозі 30-60 мг / кг / сут
(1000-3000 мг / добу) в 2 прийоми.
При недостатній ефективності призначають політерапію: вальпроати + сукцініміди (при резистентних абсансах); вальпроати + топирамат або леветірацетам (при резистентних ГСП); вальпроати + бензодіазепіни (при вираженій фотосенсітівності). Препарати карбамазепіну протипоказані.
Повна медикаментозна ремісія досягається у 85-95% хворих, причому в більшості випадків при використанні монотерапії. Проблема полягає у високій частоті рецидивів після скасування АЕП. Скасування препаратів, навіть через 4-5 років повної клінічної ремісії, викликає
рецидив нападів не менше ніж у 50% хворих. Рекомендується поступова відміна АЕП не раніше ніж через 4 роки відсутності нападів.
14.4. Епілептичні енцефалопатії дитячого та дитячого віку
синдром Веста - симптоматична або криптогенная форма генералізованої епілепсії, що характеризується нападами інфантильних спазмів, гіпсарітмія на ЕЕГ, затримкою психомоторного розвитку. Захворювання дебютує на 1-му році життя, переважно у віці 6-8 міс. Основний тип нападів - флексорние інфантильні спазми ( «салаамови напади»): дитина згинає голову і тулуб, піднімає і згинає руки і ноги. Напади дуже короткі, секундні; часто групуються в серії - до 100 і більше спазмів за 1 серію. У добу у хворих спостерігається до 10-50 серій з почастішанням після пробудження. У деяких випадках можлива виражена асиметрія спазмів, в інших - розгинання тулуба і кінцівок (екстензорная тонічні спаз- ми). Нерідко спостерігаються виражена затримка психомоторного розвитку і тетрапарез. У симптоматичних випадках зміни в неврологічному статусі виявляються незабаром після народження; при кріптогенних - тільки з початком нападів.
ЕЕГ характеризується дифузійної нерегулярної високоамплітудними повільнохвильової активністю зі слабозаметний спайковую ком- компонентом - гіпсарітмія. Можлива асиметрія епілептиформних патернів і переважання їх в потиличних відведеннях (рис. 14.3).
При нейровізуалізації визначаються дифузна атрофія, пороки розвитку головного мозку, наслідки перинатальної енце- фалопатіі. В якості окремої причини розвитку захворювання виділяють туберозний склероз, а також деякі наследственнодегенератівние і метаболічні захворювання.
Необхідно раннє призначення препарату при інфантильних спазмах. Стартова терапія починається з вігабатрин (Сабрі) - 50-100 мг / кг / добу або вальпроатов - 50-100 мг / кг / сут. Препаратом другого або третього вибору може бути топирамат (топамакс) в дозі 5-10 мг / кг / сут. При резистентних нападах призначають комбінацію зазначених базових АЕП з бензодіазепінами (клоназепам 0,25-2 мг / сут, клобазам 1 мг / кг / добу) або фенобарбіталом (5-15 мг / кг / добу), а також з суксілеп (15- 30 мг / кг / добу). При асиметричних нападах може бути доданий карбамазепін (финлепсин, тегретол) в дозі 10-20 мг / кг / сут.
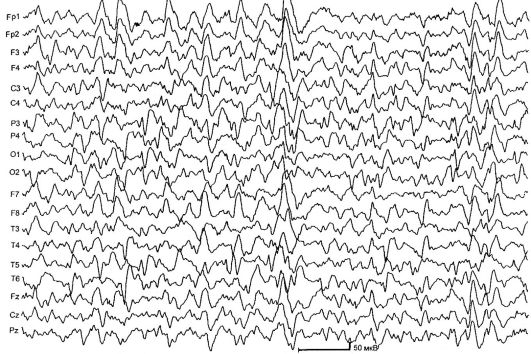
Мал. 14.3.ЕЕГ при синдромі Веста (гіпсарітмія)
Альтернативним методом є застосування кортикостероїдних гормонів (синактен-депо внутрішньом'язово; дексаметазон, перед- нізолон перорально) і імуноглобулінів (октагам). Середня дозування преднізолону - 1-2,5 мг / кг / добу з подальшим переходом на мінімальну підтримуючу дозу. Гормони призначають зазвичай в поєднанні з базовими АЕП. Лікування стероїдами проводять фахівці в клініці через загрозу розвитку важких побічних ефектів.
Прогноз складний. Сучасні АЕП дозволяють купірувати напади у 60% хворих, проте в більшості випадків залишаються виражений інтелектуальний дефект і аутистичного подібну поведінку. При персистуванні нападів спостерігається трансформація в важку мультифокальну епілепсію або синдром Леннокса-Гасто.
Синдром Леннокса-Гасто (дитяча епілептична енцефалопатія з дифузними повільними пік-хвилями на ЕЕГ) (СЛГ) - криптогенная (симптоматична) генералізована епілепсія, що характеризується частими поліморфними нападами, специфічними змінами на ЕЕГ, зниженням інтелекту, резистентністю до терапії. Етіологія в більшості випадків невідома. СЛГ - одна з найбільш важких форм епілепсії.
Захворювання дебютує частіше у віці від 3 до 8 років. Характерна тріада нападів, яка спостерігається практично в 100% випадків: тонічні аксіальні, атипові абсанси і напади падінь. Тонічні напади проявляються коротким інтенсивним напругою мускулатури тулуба і кінцівок, виникають частіше в нічний час. Іноді вони більш тривалі, супроводжуються легкими клонічними посмикуваннями кінцівок (тоніко-вібраторні напади) і вираженими вегетативними симптомами (апное, брадикардія). Атипові абсанси характеризуються більш поступовим початком і закінченням нападів, ніж при типових абсансах; свідомість нерідко флюктуирует; спостерігаються атонические феномени (падіння голови на груди, опускання плечей, нахил тулуба, подкашивание ніг). Напади падінь можуть носити різкий тонічний характер ( «падіння статуєю») або більш плавний - міатоніческій (початковий міоклонічний компонент, потім - атонія). Під час цих падінь діти отримують різні пошкодження голови і тулуба. У деяких випадках спостерігаються миоклонические та генералізовані судомні напади; поява фокальних нападів - предмет дискусії. Характерна висока частота нападів з наростанням уві сні, при пробудженні, в період пасивного неспання. Навпаки, активне неспання сприяє урежению нападів [ «мозкова активність - є антагонізм нападів» (Гасто)]. У хворих СЛГ висока ймовірність виникнення серійних нападів і епілептичного статусу (тонічні напади і атипові абсанси). Статус тонічних нападів може становити безпосередню загрозу життю пацієнтів.
У неврологічному статусі визначаються дифузна м'язова гіпотонія, атаксія. Симптоми ураження пірамідних шляхів, як правило, відсутні. Інтелект знижений у всіх випадках; може спостерігатися гіперактивний, аутістікоподобное або психопатоподібні поведінку.
На ЕЕГ виявляються 3 основних патерну: уповільнення основний активності фонової записи, повільні дифузійні комплекси гостра-повільна хвиля, пробіги швидкої (10-20 Гц) активності, частіше уві сні (рис. 14.4).
Нейровізуалізація не виявляється локальних структурних дефектів мозку; в більшості випадків визначається дифузна кортикальная атрофія.
Лікування представлено в табл. 23.
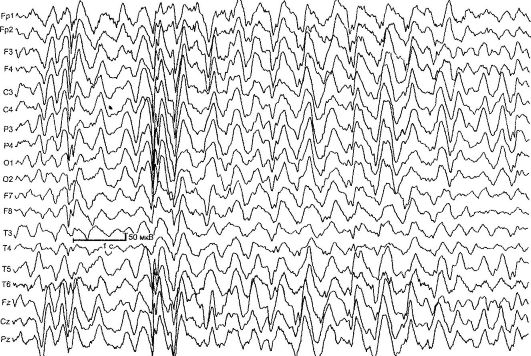
Мал. 14.4.ЕЕГ при синдромі Леннокса-Гасто
Таблиця 23.Лікування синдрому Леннокса-Гасто
Антиепілептичні препарати займають провідне місце в лікуванні СЛГ; всі інші методи - допоміжні. Стартова тера- пія починається з топирамата. Його початкова доза зазвичай становить
12,5 мг / сут. Для уникнення можливих побічних ефектів показано повільне титрування дози - збільшення на 12,5 мг щотижня. Дозування топирамата становлять 75-350 мг / сут (3-10 мг / кг / добу) і вище в 2 прийоми. Препарат другого вибору - вальпроєва кислота. Препарати вальпроєвої кислоти призначаються з поступовим збільшенням до 900-2500 мг / добу (40-80) мг / кг / сут і вище до максимально переносимої дози.
При недостатньому ефекті монотерапії (у більшості випадків) рекомендований перехід на комбінацію препаратів: топирамат + вальпроати, вальпроати + сукцініміди, вальпроати або топірамат + ламотриджин. Сукцініміди застосовують в дозі 500-1000 мг / добу (20-35 мг / кг / добу) в 3 прийоми. Ламотриджин починають з 12,5 мг / сут, нарощуючи дозу по 12,5 мг 1 раз на тиждень; середні дозування препарату - 75-200 мг / сут (3-7 мг / кг / добу) в 2 прийоми.
При резистентних до лікування тонічних нападах до базових АЕП можливе додавання карбамазепіну. У цих випадках оптимальним схема - вальпроати + карбамазепін. Карбамазепін слід призначати в невеликих або середніх дозах і тільки в комбінації з базовими АЕП. Середні дозування карбамазепіну - 100-600 мг / добу (10-20 мг / кг / добу) в 2 прийоми. Леветірацетам в дозуванні 1000- 3000 мг / добу (30-60 мг / кг / добу) може бути ефективний при міоклонічних і генералізованих судомних нападах. При переважанні тонічних нападів можлива комбінація вальпроатов і гідан- тоін. Застосовується дифенин в дозі 75-200 мг / сут (3-7 мг / кг / добу) в 2 прийоми.
При відсутності ефекту від проведеної терапії в схему лікування можливе введення бензодіазепінів в комбінації з базовими АЕП. Серед бензодіазепінів лише клобазам може бути застосований для тривалого лікування хворих СЛГ. Клобазам вводиться в дозі 10 30 мг / добу (0,5-1,0 мг / кг / добу). Всі інші бензодіазепіни повинні призначатися перорально лише як «пожежні препарати» при неконтрольованому серійному учащении нападів.
Найбільш часта комбінація при резистентних нападах у хворих СЛГ - топирамат + вальпроати + сукцініміди (або клобазам).
прогнозпри СЛГ несприятливий. Лише у 5-15% хворих вдається досягти ремісії. В інших випадках терапія сучасними АЕП дозволяє знизити частоту нападів, уникнути виникнення епілептичного статусу і зменшити інтелектуально-мнестичних
дефіцит. Тривалість життя залежить від догляду за пацієнтами. Більшість хворих - глибокі інваліди, які не здатні до самостійного життя.
Синдром Ландау-Клеффнера [придбана епілептична афазія (СЛК)] - імовірно ідіопатична форма епілепсії. Вперше електро-клінічна картина захворювання була описана В. Ландау і Ф. Клефнером в 1957 р Це досить рідкісна форма епілепсії дитячого віку, що виявляється придбаної сенсомоторної афазією в поєднанні з різними епілептичними нападами і дифузними змінами на ЕЕГ. СЛК проявляється у віці 3-7 років. До моменту дебюту захворювання рухове, психічний і мовний розвиток пацієнтів відповідає віку.
Мовні порушення - кардинальний ознака захворювання. Вони частіше розвиваються поступово, протягом декількох тижнів або місяців, рідше - катастрофічно швидко, за кілька днів. Перший симптом захворювання, як правило, однотипний: батьки відзначають, що дитина перестає адекватно реагувати на звернену мову (прояви сенсорної афазії). У цей період можуть з'явитися виражені порушення поведінки: емоційна лабільність, збудливість, гіперактивність; відзначаються негативізм, спалахи агресії. Надалі виникають порушення експресивної мови: пацієнти починають говорити простими фразами, потім вживають лише окремі слова і перестають говорити взагалі.
Другий симптомокомплекс СЛК - епілептичні напади. Характерні фокальні моторні напади (фарінгооральние і геми- фаціальні), а також атипові абсанси. Рідше зустрічаються атонические, міоклонічні та генералізовані судомні пароксизми. У більшості випадків напади рідкісні; спостерігаються при засипанні і пробудженні. У 1/4 хворих епілептичні напади відсутні. У цих випадках діагноз встановлюється на підставі виникнення придбаної афазії, виражених когнітивних порушень і даних ЕЕГ.
У неврологічному статусі осередкові симптоми відсутні. При психологічному тестуванні виявляють сенсорну або тоталь- ву афазію, порушення праксису. Характерні розлади поведінки.
ЕЕГ визначає наявність епілептиформних порушень в 100% випадків. Типові високоамплітудні (200-400 мкВ) регіональні гострі хвилі або комплекси гостра-повільна хвиля, локалізованих
ванні переважно в задневісочних або тім'яно-скроневих областях. Епілептиформна активність наростає уві сні (в фазі як швидкого, так і повільного сну), поширюється дифузно, зазвичай зберігаючи амплітудне переважання домінантного для мови півкулі. На окремих епохах записи уві сні індекс епілептиформні активності може досягати 100%. Саме епілептиформна активність призводить до розвитку важких мовних порушень (прояв когнітивної епілептиформної дезінтеграції). МРТ, як правило, в нормі.
Схема лікування СЛК залежить від наявності або відсутності епілептичних нападів. При СЛК без епілептичних нападів ефективна монотерапія сукцініміди або бензодіазепінами. Стартове лікування здійснюється сукцініміди. Етосуксимід призначається в дозі 500-1000 мг / добу (25-35 мг / кг / добу) в 3 прийоми. Препарат другого вибору - клобазам в дозі 10-30 мг / сут (0,5-1,0 мг / кг / добу) в 2-3 прийоми. Дані препарати блокують продовжену диффузную епілептиформні активності на ЕЕГ, приводячи до вос- становлення мовних функцій. При наявності епілептичних нападів вони застосовуються тільки як додаткові АЕП.
При СЛК з епілептичними нападами лікування починають з вальпроєвоїкислоти в дозі 900-2000 мг / добу (30-70 мг / кг / добу) в 2 прийоми. Препарат другого вибору - топирамат. Топамакс призначається з поступовим збільшенням дози до 50-150 мг / сут (3-7 мг / кг / добу) в 2 прийоми. При неефективності монотерапії слід переходити до комбінованого лікування. Оптимальні комбінації при СЛК: вальпроати + сукцініміди, вальпроати + топирамат, вальпроати + бензодіазепіни. Одним з найважливіших критеріїв ефективності терапії є блокування феномена вторинної білатеральної синхронізації на ЕЕГ (дифузних розрядів).
Застосування карбамазепіну протипоказано через можливий почастішання нападів, посилення вторинної білатеральної сінхроні- зації на ЕЕГ і поглиблення мовних порушень.
Кортикостероїди (синактен-депо, дексаметазон) є препаратами резерву. Вони володіють позитивним ефектом в отноше- ванні відновлення мови. Можлива пульс-терапія дексаметазоном в дозі 1 мг / кг / сут. Метод полягає в призначенні препарату кожні 2 тижні; потім інтервал без застосування дексаметазону становить 4-8 тижнів, потім знову 2-тижневий курс. При цьому базова терапія АЕП проводиться без перерви.
В якості хірургічного лікування при СЛК застосовують субпіальние насічки.
прогнозпри СЛК щодо епілептичних нападів сприятливий: у 100% пацієнтів напади повністю купіруються до пубертатного періоду (під дією АЕП або спонтанно). Разом з тим при відсутності терапії або неадекватному лікуванні (можливо, при нерозпізнаної епілептичної природі захворювання) мовні і когнітивні порушення можуть персистувати.
Епілепсія з електричним епілептичним статусом повільного сну (Синоніми: епілепсія з безперервною пік-хвильової активністю на ЕЕГ під час повільного сну, ESES-syndrom - electrical status epilepticus during slow sleep) за класифікацією 1989 р відноситься до форм, які мають риси як генералізованих, так і парціальних. Патогенез синдрому пов'язаний з постійною «бомбардуванням» продовженої епілептиформні активністю коркових центрів з розвитком їх функціонального гальмування і розривом нейрональних зв'язків, що призводить до розвитку важких когнітивних порушень.
Патогномонічні наявність фокальних і псевдогенералізованних епілептичних нападів у поєднанні з вираженими когнітивними порушеннями і патерном продовженої дифузійної епілептиформні активності в період повільного сну, що триває постійно багато місяців і років.
Виділяють ідіопатичний і симптоматичний варіанти синдрому. При симптоматичному варіанті затримка психомоторного розвитку, осередкові неврологічні симптоми (косоокість, геміпаретична форма ДЦП, атаксія), структурні зміни при нейровізуалізації присутні до початку нападів. При «класичному» (ідіопатичному) варіанті дані ознаки відсутні. Вік дебюту епілептичних нападів варіює, по спостереженню Тассінарі (2002), від 8 місяців до 12 років, складаючи в середньому 4,7 року. Серед хворих переважають хлопчики. Не менш ніж у 1/3 пацієнтів епілептичні напади відсутні. При цьому діагноз встановлюється на підставі поєднання постійної продовженої епілептиформні активності в повільному сні з вираженими когнітивними порушеннями.
Характерно початок захворювання з фокальних моторних (фарінгооральних, геміфаціальний, унилатеральной) нападів або аль- тернірующіх геміконвульсіі, що виникають переважно у
час сну (особливо - перед пробудженням). У 15% випадків в анамнезі констатують фебрильні судоми. Напади, як правило, рідкісні; в деяких випадках - поодинокі. На даному етапі ще немає виражених порушень когнітивних функцій. У цей період захворювання діагноз не може бути встановлений.
Другий період (розгорнутих клінічних проявів) настає через кілька місяців або років з моменту дебюту перших нападів. Клінічно він характеризується появою «псевдогенералізованних» нападів і, перш за все, атипових абсансов, зазвичай з атоническим компонентом ( «кивки», нахили тулуба вперед, підгинання ніг). Крім того, можливі міоклонічні напади, пароксизми падінь і генералізовані тоніко-клонічні напади. Більшість даних нападів - результат феномена вторинної білатеральної синхронізації на ЕЕГ. З появою цього феномена стають помітними і швидко наростають когнітивні порушення. Розлад когнітивних функцій (пам'яті, уваги, швидкості реакції, виконання команд та ін.) З порушенням соціальної адаптації та неможливістю навчання називається «дитячої епілептиформної когнітивної дезінтеграцією». Змінюється поведінка (псіхопато-, шізофрено-, аутістікоподобний синдроми). Порушення мови включають сенсорну або моторну афазію, оролінгвобуккомоторную діспраксії, слухову агнозію. Виникає стійкий гемипарез або атаксія (при розташуванні епілептичного вогнища переважно в моторній корі). До рідких симптомів відносять Алексію, акалькулія. У більшості випадків всі типи порушень в тій чи іншій мірі поєднуються. Поява в клініці захворювання «псевдогенералізованних» нападів і порушення вищих психічних функцій корелює з виникненням на ЕЕГ продовженої епілептиформні активності в повільному сні.
На третьому, заключному етапі частота нападів поступово знижується; вони стають рідкісними, поодинокими, більш чутливими до терапії. При цьому відбувається поступове неухильне поліпшення вищих психічних і рухових функцій (зазвичай з початком пубертатного періоду).
ЕЕГ грає вирішальну роль в діагностиці ЕЕСМ. Можливо відсутність епілептиформні активності в період неспання. Характерно поява і різке наростання дифузійної епілептиформні активності в період повільного сну з найвищим її індексом, що досягає 85-100% в цю фазу. Ця діяльність
триває постійно багато місяців і років. Фізіологічні патерни сну зникають. У період REM-сну епілептиформна актив ність зменшується або блокується.
Методи нейровізуалізації в більшості випадків порушень не виявляють. При симптоматичних варіантах відзначаються локальні порушення, що виникають внаслідок перинатального ураження, дисгенезии мозку.
Тактика лікування залежить від наявності або відсутності епілептичних нападів при синдромі ЕЕСМ. При електричному епі- лептіческіе статус повільного сну без епілептичних нападів ефективна монотерапія сукцініміди або бензодіазепінами. Етосуксимід призначається в дозі 500-1000 мг / добу (25-35 мг / кг / добу) в 3 прийоми. Препарат другого вибору - бензодіазепіни. Клобазам застосовується в дозі 10-30 мг / сут (0,5-1,0 мг / кг / добу) в 2-3 прийоми. Дані препарати різко блокують продовжену диффузную епі- лептіформную активність на ЕЕГ і опосередковано призводять до поліпшення когнітивних функцій.
При наявності епілептичних нападів вони застосовуються тільки як додаткові АЕП, а стартова терапія здійснюється препаратами вальпроєвої кислоти, а потім топираматом у вигляді монотерапії. Вальпроати призначаються в дозі 600-2000 мг / добу (30-70 мг / кг / добу) в 2 прийоми. Препарат другого вибору - топирамат, призначається з поступовим збільшенням дози до 50-150 мг / сут (3-7 мг / кг / добу) в 2 прийоми.
При недостатній ефективності монотерапії застосовується комбіноване лікування. Оптимальні комбінації: вальпроати + сукцініміди, вальпроати + топирамат, вальпроати + бензодіазепіни (клобазам). Найважливіший критерій ефективності лікування - зменшення індексу або повне блокування продовженої епілептиформні активності на ЕЕГ. Застосування карбамазепіну протипоказано через можливість появи або почастішання нападів, а також посилення вторинної білатеральної синхронізації на ЕЕГ і поглиблення когнітивних порушень.
У резистентних випадках до базових АЕП доводиться додавати кортикостероїди (синактен-депо, преднізолон, метипред, дексаметазон і ін.). Синактен-депо призначається, починаючи з 0,1 мг / сут, зі збільшенням поступово по 0,1 мг раз в 3-5 днів до 1,0 мг / добу. Тривалість лікування становить від 3-4 тижнів до декількох місяців з поступовою відміною. При цьому базова терапія АЕП
проводиться без перерви. Гормони мають виражений блокуючим ефектом відносно епілептиформні активності на ЕЕГ і сприяють поліпшенню мовних функцій.
У разі симптоматичного характеру можливо хірургічне лікування - кортикальна резекція. Наприклад, при гемімегален- Цефалий єдиним методом уникнення важкої незворотною когнітивної дезінтеграції є функціональна гемісферотомія.
Прогноз сприятливий щодо епілептичних нападів і серйозний для когнітивних порушень. Напади добре відповідають на адекватну терапію АЕП і зазвичай зникають після 10-12 років. З поступовим зникненням продовженої епілептиформні активності в фазу повільного сну покращуються і когнітивні функції до настання пубертатного періоду. Однак половина всіх пацієнтів «виходить» з захворювання з вираженим інтелектуально-мнестичних дефектом і не здатна до навчання в загальноосвітній школі.
Специфічні синдроми. Серед специфічних синдромів в педіатричній неврології особливо актуальні фебрильні судоми і епілептичний статус.
14.5. фебрильні судоми
фебрильні судоми (ФС) - судоми у дітей у віці від 6 місяців до 5 років, що виникають при температурі, не пов'язаної з нейроинфекцией. Судоми у дітей в гострій стадії менінгіту, енцефаліту не належать до категорії ФС, а розглядаються як симптоматичні прояви нейроінфекції. ФС зустрічаються у 5% дітей. В основі ФС лежить генетично детерміноване зниження порога судомної готовності з виникненням генералізованих судомних розрядів при гіпертермії. Спадкування полигенное; передбачаються дефекти в локусі FEB1 (хромосома 8ql3-q21) і FEB4 на хромосомі 5ql4ql5. Імовірність появи ФС у дітей, якщо у одного з батьків вони були в анамнезі, може бути досить високою - 5-20%.
Виділяють типові (прості) і атипові (складні) ФС. Типові ФС генетично детерміновані, виникають у неврологіче- скі здорових дітей і складають до 90% всіх випадків ФС. Виявляються генералізованими тоніко-клонічними нападами на тлі високої лихоманки. ФС, як правило, виникають в 1-й день підвищення температури, зазвичай, коли дитина засинає. тривалість
нападів не перевищує 10 хв, постпріступном симптоми випадання відсутні. ЕЕГ в межприступном періоді в межах норми. Більш ніж у 50% дітей ФС повторюються. Типові ФС не впливають на розвиток дитини і безслідно проходять без лікування після 5 років. Ризик трансформації в епілепсію (переважно идиопатические форми) становить не більше 10%.
Атипові ФС (складні) складають близько 10% випадків всіх ФС. Вони характеризуються такими ознаками
Вік дебюту до 1 року або після 5 років.
Висока тривалість нападів - більше 30 хв.
Перший напад може проявлятися епілептичним статусом, що вимагає реанімаційних заходів.
Напади з фокальним компонентом: адверсія голови і очей, геміконвульсіі, «обмяканіе».
Виникнення симптомів випадання після нападу (наприклад, тоддовскіе паралічі, афазія).
Виявляються осередкові неврологічні симптоми і когнітивні порушення.
Регіональне уповільнення на ЕЕГ по одному з скроневих відведень.
На МРТ визначається грізна ознака атипових ФС - мезіальний скроневий склероз (результат ішемічного інсульту при тривалих ФС, нашаровуються на незрілий мозок).
Атипові ФС мають несприятливий прогноз. Вони часто поєднуються з когнітивними порушеннями. Ризик трансформації в епі- Лепсе становить близько 15%; при наявності мезиального височного склерозу - різко підвищується. Тривалі Некупейні атипові ФС можуть призводити до розвитку гострого порушення мозкового кровообігу за ішемічним типом і появи стійкого моторного дефіциту. У цьому випадку після епізоду ФС розвиваються гемипарез і резистентна епілепсія з геміконвульсівнимі нападами: синдром ННЕ (епілепсія з геміконвульсіі і гемиплегией).
Хворі з типовими ФС не потребують тривалої терапії та медикаментозної профілактики. Рекомендується фізичне охолодження при гіпертермії (провітрювання, розтирання спиртом, оцтом), введення літичних сумішей. При повторних ФС рекомендується навчити батьків вводити препарати діазепаму в дозі 0,5-1,5 мл внутрішньом'язово в момент нападу. Це робиться для профілактики розвитку тривалого нападу і епілептичного статусу. терапія
АЕП не проводиться. В окремих випадках можливе профілактичне призначення бензодіазепінів або фенобарбіталу в терапевтичній дозі на період лихоманки (3-5 днів). Однак ефективність такої «профілактики» не доведена.
У разі встановлення діагнозу атипових ФС, навпаки, необхідно призначати лікування, як при епілепсії (наприклад, препара- ти карбамазепіну або вальпроати в вікових дозуваннях). При важкому тривалому нападі атипових ФС здійснюються ті ж заходи, що і при епілептичному статусі.
Диференціальний діагноз епілепсії проводять з синкопальними станами, психогенними порушеннями, розладами сну, Неепілептіческіе міоклонусом, мігренню, гіперкінезами. У клінічній практиці найбільші труднощі викликає диференціальна діагностика епілепсії з синкопальними станами і конверсійними (психогенними) нападами.
14.6. Загальні принципи лікування епілепсії
Основний принцип: максимум терапевтичної ефективності при мінімумі побічних ефектів. Хворі, які страждають на епілепсію, змушені застосовувати АЕП протягом багатьох років. У зв'язку з цим важливою вимогою до проведеної терапії є відсутність негативного впливу препаратів на якість життя пацієнтів.
Хворий повинен дотримуватися режиму сну і неспання; уникати недосипання, пізнього відходу до сну і раннього (особливо раптового) пробудження. Підліткам і дорослим пацієнтам рекомендується віз триматися від прийому спиртних напоїв. Слід уникати впливу ритмічної светостімуляціі при формах епілепсії з вираженою фотосенсітівная. Чітке дотримання цих правил дозволяє знизити частоту епілептичних нападів у 20% пацієнтів.
Лікування епілепсії може бути почато тільки після встановлення точного діагнозу. Профілактичне лікування епілепсії неприпустимо! Зміни на ЕЕГ при відсутності будь-яких симптомів забо- левания не є приводом для призначення терапії. Виняток становить важка черепно-мозкова травма (забиття головного мозку, виникнення гематоми), після якої можливе призначення базового АЕП терміном на 6-12 міс. При епілептичних енцефалопатії лікування може бути призначено при відсутності нападів на підставі поєднання дифузійної епілептиформні активності з вираженими порушеннями вищих психічних функцій.
Лікування епілепсії слід починати після повторного нападу. Одиничний пароксизм може бути «випадковим», обумовленим лихоманкою, метаболічними розладами і не ставитися до епілепсії. АЕП призначають тільки в разі повторних непровоціруемих епілептичних нападів. При деяких доброякісних епілептичних синдромах дитячого віку (насамперед РЕ) і рефлекторних формах епілепсії (епілепсія читання, первинна фотосенсітівная епілепсія) допускається ведення пацієнтів без АЕП, якщо напади дуже рідкісні і легко протікають при проведенні превентивних заходів.
При призначенні АЕП важливо враховувати принцип монотерапії: стартове лікування здійснюється одним препаратом. Монотерапія дозволяє уникнути виникнення важких побічних ефектів і тератогенного впливу, частота яких значно зростає при призначенні кількох препаратів одночасно. Виняток становлять резистентні форми епілепсії (синдром Веста, синдром Леннокса-Гасто, симптоматичні фокальні епілепсії), при яких неможливо домогтися ефекту без застосування комби- лося терапії. Препарати призначають строго відповідно до форми епілепсії і характером нападів. Успіх лікування епілепсії багато в чому визначається точністю сіндромологіческой діагностики.
Вперше препарат призначають, починаючи з малої дози, поступово збільшуючи її до досягнення терапевтичного ефекту або появ- лення перших ознак побічних явищ. При цьому враховують ефективність і переносимість препарату, а не зміст його в крові. Стартова доза зазвичай становить 1/8 - 1/4 від передбачуваної середньої терапевтичної. Збільшення дозування відбувається раз в 5-7 днів (в залежності від переносимості препарату та особливостей перебігу епілепсії).
Необхідно призначати АЕП в адекватних вікових дозуваннях. Застосування малих доз - одна з основних причин «псевдорезістентной» в клінічній практиці. Слід пам'ятати, що при важких формах епілепсії єдиний шанс реально допомогти пацієнтові - призначати АЕП в високих дозах (табл. 24).
У разі неефективності препарату його поступово замінюють іншим, потенційно ефективним при даній формі епілепсії. Не можна відразу додавати другий препарат, тобто переходити на полі терапію.
Існує близько 30 АЕП з різним спектром антиепілептичної активності і побічних ефектів. Перевагу віддають сучасним АЕП, які мають широкий спектр клінічної ефективності і добре стерпним (вальпроати, топірамат). Рекомендується лікування пролонгованими препаратами, які призначаються 2 рази на добу (конвулекс ретард, депакин-хроно, фінлепсин ретард, тегретол CR). До базових АЕП відносять вальпроати (депакин, конвулекс, КОНВУЛЬСОФІНУ) і карбамазепін (финлепсин, тегретол, три- лептал). Сукцініміди (суксилеп), бензодіазепіни (клоназепам, клобазам) і ламотриджин (ламіктал) застосовуються у дітей у вигляді додаткової терапії. Нові АЕП (топірамат, леветірацетам, окскарбазепін) призначають в якості монотерапії та в комбінації з базовими АЕП. До старих АЕП відносять барбітурати (фенобарбі- тал, гексамидин, бензонал) і гідантоїни (дифенін, фенітоїн); вони мають виражені побічними ефектами і останнім часом призначаються все рідше.
Для контролю терапії і побічних ефектів необхідно 1 раз в 3 міс робити клінічний аналіз крові з обов'язковим дослідженням рівня тромбоцитів, а також біохімічний аналіз крові з визначенням змісту білірубіну, холестерину, печінкових ферментів. Раз в 6 міс проводиться УЗД органів черевної порожнини. Рекомендується також при кожному огляді контролювати рівень антиепілептичних препаратів в крові. Обов'язково ведення щоденника пацієнтом або його батьками.
Таблиця 24.Причини відсутності ефекту від призначення АЕП
У резистентних випадках показані хірургічна резекція, стимуляція блукаючого нерва і кетогенная дієта. Слід строго дотримуватися показань до хірургічного лікування, пацієнтам необхідно проводити прехірургіческое обстеження, яке можливе лише в спеціалізованих центрах. При симптоматичних фокальних формах епілепсії, резистентних до АЕП, хірургічне лікування може бути єдиною можливістю повністю позбавити пацієнтів від нападів.
стійка ремісія - відсутність нападів протягом 1 року і більше. Про повноїремісії говорять за відсутності нападів і нормалізації ЕЕГ.
Терміни зниження і скасування АЕП строго індивідуальні і залежать насамперед від форми епілепсії і особливостей перебігу захворювання. При ідіопатичних фокальних формах і дитячої абсансепілепсіі зниження АЕП може починатися після 3 років відсутності нападів; при симптоматичних фокальних епілепсія, юнацьких варіантах ідіопатичною генералізованою епілепсії - не раніше ніж через 4 роки ремісії. Повне скасування АЕП здійснюється поступово, зазвичай протягом 1 року.
Фактори несприятливого прогнозу: недоношеність в анамнезі, ранній дебют нападів, епілептичні статуси, осередкові неврологічні порушення, зниження інтелекту, виражені порушення поведінки, наявність на ЕЕГ продовженого регіонального уповільнення або феномена вторинної білатеральної синхронізації, структурні зміни при нейровізуалізації, відсутність ефекту від застосування базових АЕП в адекватних дозуваннях. В даний час завдяки застосуванню всього арсеналу АЕП в цілому у 65% хворих вдається домогтися стійкої ремісії нападів.
14.7. епілептичний статус
епілептичний статус (ЕС) - це напад тривалістю більше 30 хв або повторні часті напади, між якими свідомість повністю не відновлюється. Стану, загрозливі щодо розвитку ЕС: тривалий (понад 5 хв) напад або більше 3 генералізованих судомних нападів, що виникли протягом 24 год.
В середньому частота виникнення ЕС становить 28 випадків на 100 000 загальній популяції і 41 на 100 000 дитячого населення. У 5% дорослих хворих і 20% дітей з епілепсією в анамнезі відбувався ЕС. У 26% випадків ЕС виникає у дітей 1 року життя, в 43% випадків -
в перші 2 роки, а в перші 3 роки - в 54%. ЕС становить до 4% всіх випадків в невідкладної неврології. Смертність при ЕС при відсутності спеціалізованої допомоги становить до 50%, а при адекватному лікуванні - 5-12%.
До ЕС призводять погіршення перебігу епілепсії, неправильний прийом АЕП, інфекційні захворювання з лихоманкою. ЕС ускладнюють ЧМТ, гематома, інсульт, нейроінфекції, екзогенні інтоксикації, важкі метаболічні розлади та ін.
Патогенез ЕС включає 2 фази.
1. Триваюча судомна активність прискорює метаболізм мозку, у відповідь підвищуються церебральний кровотік і приплив кисню і глюкози. Поступово компенсаторні механізми виснажуються, розвивається ацидоз, у тканинах мозку підвищується рівень лактату. Це призводить до порушення серцевої діяльності: підвищуються артеріальний тиск, серцевий викид і частота серцевих скорочень. Підвищення активності симпатичної системи викликає поява гіперсалівації, пітливості, збільшення бронхі- альної секреції, гиперпирексии. Відзначається гіперглікемія, обумовлена підвищенням викиду адреналіну і норадреналіну.
2. Зрив компенсаторних механізмів призводить до розвитку набряку і дистрофії нейронів, подальшого посилення епілептогенеза. Церебральний кровотік починає залежати від системного артеріального тиску. Гіпотензію поглиблюють гіпоксія, деякі лікарські засоби (Наприклад, внутрішньовенне введення реланиума). Розвивається набряк мозку, знижується мозковий кровотік. Результатом усіх цих змін є ішемія мозку, гіпоксія і ацидоз. Пізніше приєднується поліорганна недостатність: системний ацидоз, гіпоглікемія, порушення функції печінки, ниркова недостатність, Рабдоміоліз, ДВС-синдром. Можливі ускладнення інтенсивної терапії: інфекції, емболія судин легенів, дисбаланс електролітів.
Протягом ЕС виділяють:
Предстатус (0-9 хв з моменту початку нападів);
Початковий (10-30 хв);
Розгорнутий (31-60 хв);
Рефрактерний (понад 60 хв).
судомний ЕС- це стан, при якому постійні або періодично перериваються тоніко-клонічні судоми зберігаються більше 30 хв без відновлення свідомості між нападами. Судомний ЕС становить 10-25% всіх випадків ЕС. Спочатку напади стають частішими або тривалими (з розвитком стану, якому загрожує по ЕС). У цей період розвиток ЕС може бути припинено. Типові тоніко-клонічні судоми з часом стають все більш частими, виникає повна втрата свідомості. У стані коми клоническая активність може зменшуватися - практично до повного зникнення. В цей час наростають респіраторні, циркуляторні та метаболічні порушення. На ЕЕГ при судорожному ЕС спостерігається генералізована епілептиформна активність у вигляді гострих хвиль, спайки, швидких спайк-хвильових комплексів з подальшим уповільненням. Біоелектрична активність маскується великою кількістю Міографічний і рухових артефактів. У другій стадії ЕС основна активність сповільнюється і ущільнюється. Поява «періодичних латералізованние епілептиформних розладів» і тріфазних хвиль спостерігається при великій тривалості ЕС і є маркером несприятливого прогнозу (летального результату або розвитку вегетативного стану). Смертність при судорожному ЕС становить 5-19% і залежить від етіології. Неврологічні і психічні порушення пропорційні тривалості статусу.
Особливою формою ЕС у дітей є геміконвульсівно-Геміплегічна епілептичний синдром.Він виникає у дітей перших 4 років життя, частіше при лихоманці, і характеризується важким тривалим статусом генералізованих судомних нападів з виразним одностороннім акцентом. Після закінчення статусу у пацієнта відзначається тривала геміплегія, що виникає на стороні переважання судом. У більшості випадків прогноз поганий - в подальшому у 85% дітей розвивається резистентна до лікування симптоматична фокальна епілепсія з інтелектуально-мнестичних розладами і моторним дефіцитом.
ЕС при епілепсії Кожевнікова проявляється постійними миоклоническими нападами, обмеженими певним сегментом тулуба. Періодично можуть виникати моторні джексоновские напади з вторинною генералізацією.
ЕС миоклонических нападів проявляється неконтрольованими частими, практично безперервними миоклониями, більш вираженими в дистальних відділах кінцівок, і супроводжується
оглушення, а не повною втратою свідомості. Миоклонические епілептичний статус протікає підступно, виникає поволі і може тривати протягом декількох днів, місяців і навіть років, супроводжуючись прогресуючою деменцією. ЕЕГ при міоклонічних статус зазвичай виявляє множинні поліспайк-хвильові розряди на тлі відсутності фізіологічної фонової активності, а також дифузне продовжене уповільнення, що чергується з множинними мультифокальних спайками і дифузними і генералізованими спайк- і поліспайк-хвильовими комплексами.
бессудорожная ЕС (Статус абсансов) характеризується появою регулярної генералізованої пік-хвильової активності на ЕЕГ. Найбільш типовим видом статусу абсансов в дитячому віці є ЕС типових абсансов (пік-хвильової ступор). Найчастіше він спостерігається в рамках дитячої та юнацької абсансной епілепсії, рідше - при юнацької міоклонічні епілепсії. Проявляється різким почастішанням абсансов, наступних один за іншим безпосередньо або з дуже коротким інтервалом. Виникають амимия, слинотеча, ступор. Дитина виглядає мрійливим, руху уповільнені. Ступінь порушення свідомості варіює. Діти іноді зберігають можливість реагувати на оклик і виконувати прості завдання. Можуть визначатися миоклонии м'язів обличчя, плечей, рук. Тривалість статусу - від декількох хвилин до декількох годин і навіть діб. Статус абсансов частіше виникає в ранкові години, безпосередньо після пробудження пацієнтів і нерідко закінчується генералізованим судорожним нападом. У половини хворих статус рецидивує при неадекватному лікуванні. Статус абсансов провокується недосипанням або неправильним лікуванням, зокрема застосуванням карбамазепіну і вігабатрин.
ЕС складних фокальних нападів клінічно вариабелен. Зазвичай він починається періодом спутаного свідомості, що триває від декількох годин до декількох днів. Очі широко розкриті, обличчя гипомимично. Спостерігаються тривалі амбулаторні автоматизми, що зовні нагадують цілеспрямовані, доцільні і координовані рухи, зазвичай з взаємодією. У цьому стані хворі можуть безцільно блукати вулицями; сідають в транспорт, їдуть в інші міста. Часто свідомість вимкнено в повному обсязі, і може зберігатися частковий контакт з пацієнтом. Можливі сприятливі фактори включають прийом алкоголю, лікарську залежність, інфекції, менструації, електросудо-
рожнов терапію. На ЕЕГ спостерігається постійна або періодична, регіональна (частіше в скроневих відведеннях) пароксизмальна активність у вигляді комплексів пік-хвиля, ізольованих піків або повільної пік-хвильової активності.
Лікування епілептичного статусу. У початкових стадіях доцільно використовувати швидкодіючі препарати, а на більш пізніх - препарати, які не накопичуються в організмі і мають мінімум побічних ефектів. Лікувальні заходи при ЕС строго диференційовані в залежності від стадії ЕС: в 1-й стадії лікувальні заходи виконуються на догоспітальному етапі; у 2-й і 3-й - в умовах палати інтенсивної терапії неврологічного відділення в 4-й - в реанімаційному відділенні. У 2-й стадії необхідне проведення всіх діагностичних заходів для виявлення етіології ЕС і моніторингу показників життєво важливих функцій.
I. Перед статус (0-9 хв з моменту початку нападів) - своєчасно почате адекватне лікування може запобігти розвитку важкого ЕС:
Забезпечення прохідності дихальних шляхів;
оксигенотерапія;
Діазепам (в 2 мл 10 мг) в / в 0,25 мг / кг, швидкість введення - 2-4 мг / хв. Можливо неодноразове повторення кожні 30 хв. Сумарна доза препарату на добу не повинна перевищувати 40 мг. Основний побічний ефект - пригнічення дихання.
II. Ранній статус (10-30 хв):
Діазепам продовжувати;
Лоразепам (в 1 мл 4 мг) 0,05-0,1 мг / кг зі швидкістю 2 мг / хв. Вводиться 1 або 2 рази з інтервалом в 20 хв, сумарно - не більше 4 мг. Побічні ефекти: розвиток толерантності після 1-2 ін'єкцій; рідко - пригнічення дихання (менш виражене, ніж при застосуванні діазепаму), артеріальна гіпотензія;
Фенітоїн (діфантоін) (в 5 мл 250 мг) в / в, розвести в фізіологічному розчині 5-20 мг / мл. Дозування - 15-20 мг / кг зі швидкістю 25 мг / хв. Можливо повторне введення препарату кожні 6 год у дозі 5 мг / кг в / в або перорально через зонд. Концентрація фенітоїну в крові повинна підтримуватися на рівні 20-25 мкг / мл.
Побічні ефекти: зупинка серця, артеріальна гіпотензія, флебосклероз. При відсутності фенітоїну можливе введення оксибутират натрію (ГОМК) (в 1 мл 20% розчину 200 мг) в / в. Дозування - 100-150 мг / кг зі швидкістю 400 мг / хв. Побічний ефект - гіпокаліємія.
III. Розгорнутий статус (31-60 хв):
Діазепам або лоразепам;
Фенобарбітал (в 1 мл 200 мг) в / в. Дозування дітям до 1 року - 20 мг / кг, далі - 15 мг / кг зі швидкістю до 100 мг / хв. Разова доза не повинна перевищувати максимальну вікову або бути більше 1000 мг. Можливо введення препарату кожні 8 год в дозі 3-5 мг / кг / добу перорально через зонд. Побічні ефекти: зниження скорочувальної здатності міокарда, пригнічення дихання, пригнічення свідомості, артеріальна гіпотензія;
Альтернативою є в / в введення депакина для внутрішньовенних ін'єкцій у дозі 20-25 мг / кг перші 5-10 хв, далі - 2 мг / кг / год. Стандартна доза - 25 мг / кг / сут. Підтримуюча доза 5 мг вводиться 4 рази на добу або проводиться постійна інфузія в дозі 1 мг / кг / год. Депакін для внутрішньовенного введення не пригнічує дихання і серцеву діяльність; швидко досягається необхідна концентрація в плазмі крові; дозволяє уникнути інтубації хворого; високоефективний (80-90%), в тому числі при неефективності діазепаму і фенітоїну; гарантує відсутність рецидиву нападів протягом 24 год.
IV. Рефрактерний статус (понад 60 хв) супроводжується важкими, нерідко незворотними змінами в головному мозку і внутрішніх органах, Розладами метаболізму:
Інтубірованіе пацієнта з перекладом на штучну вентиляцію легенів в реанімаційному відділенні;
Барбітуровий наркоз: введення тіопенталу натрію (в 1 мл 2,5% розчину 25 мг) в / в в середній дозуванні 100-250 мг протягом 20 с. При відсутності ефекту - додаткове введення препарату в дозі 50 мг в / в кожні 3 хв до повного купірування нападів. Далі перехід на підтримуючу дозу - в середньому 3-5 мг / кг в / в кожну годину (необхідний постійний моніторинг рівня препарату в крові). Сумарна доза препарату не повинна перевищувати 1 г. Тривалість барбитурового наркозу зазвичай становить 12-24 год. Ускладнення: зниження скорочувальної здатності міокарда, важке пригнічення дихання,
артеріальна гіпотензія, токсичний гепатит і панкреатит, анафілактичний шок;
Після ліквідації ЕС і при відновленні свідомості - перехід на пероральний прийом необхідних антиепілептичних препаратів.
Під час 2-4-й стадій ЕС проводиться додаткова терапія, спрямована на корекцію життєво важливих функцій, електролітнихпорушень, Боротьбу з набряком мозку (дексаметазону натрієва сіль 4 мг в / в кожні 6 год або манітол 1,0-1,5 г / кг в / в крапельно зі швидкістю 60-80 крапель / хв).
Для лікування епілептичного статусу у дітей 1 року життя застосовують:
бензодіазепіни
Діазепам (0,5 мг / кг per rectum, в / м або в / в),
Лоразепам (0,2 мг / кг per rectum або в / в),
Мідазолам (0,15-0,4 мг / кг в / в болюсно, підтримуюча інфузія - 1-3 мкг / кг / хв);
гідантоїни
Фосфенітоїн (20 мг / кг в / в),
Фенітоїн (20 мг / кг в / в, максимальна швидкість введення - 25 мг / хв, концентрація в плазмі - 20-25 мкг / мл).
При рефрактерном статус застосовуються:
Оксибутират натрію (ГОМК) в дозі 100-150 мг / кг зі швидкістю 400 мг / хв;
Фенобарбітал (20 мг / кг в / в, повторно болюсно через 20-30 хв, максимальна доза - 100 мг / кг на добу);
Пропофол (3 мг / кг в / в болюсно, потім інфузія 100 мкг / кг / хв).
Ін'єкційний депакин 400 мг в 1 флаконі: початкова доза - 15-25 мг / кг, потім підтримуюча інфузія - 1-4 мг / кг / год.
Прогноз.Наслідками ЕС можуть бути: повне одужання, Виздо- ровленіе з наявністю стійких порушень, летальний результат. В цілому ризик розвитку різних ускладнень тим вище, чим молодша дитина. У багатьох хворих після судомного ЕС на КТ або МРТ виявляють дифузну або локальну атрофію кори великого мозку. До застосування сучасних методів лікування на початку ХХ ст. смертність від ЕС становила 51%; в кінці ХХ ст. -18%. Смертність вище при ЕС на тлі інсульту, енцефаліту, черепно-мозкової травми і пухлини головного мозку. Летальний результат вкрай рідкісний при ЕС в рамках ідіопатичною генералізованою епілепсії (не більше 1% випадків).
1. Карлов В.А.Судомний епілептичний статус: вирішена і невирішену // Неврологічний журнал. - 2000. -? 3. - С. 4-8.
2. Литвинович ЕФ, Савченко А.Ю., посполите А.В.Сучасні аспекти фармакотерапії епілептичного статусу // Клінічна епілептології. - 2007. -? 1. - С. 28-32.
3. Мухін К.Ю., Петрухін А.С.Идиопатические форми епілепсії: систематика, діагностика, терапія. - М .: Медицина, 2000. -
319 с.
4. Петрухін А.С.Епілептології дитячого віку. - М .: Медицина,
2000. - 623 с.
5. Aicardi J., Chevrie J.J.Convulsive status epilepticus in infants and children // Epilepsia. - 1970. - Vol. 11. - Р. 187-197.
6. Aminoff M.J., Simon R.P.Status epilepticus: causes, clinical features, and consequenses in 98 patients // Am. J. Med. - 1980. - Vol. 69. -
Р. 657-666.
7. Brown J.K., Hussain N.H.Status epilepticus 1: pathogenesis // Dev. Med. Clin. Neurol. - 1991. - Vol. 33. - P. 3-17.
8. Cascino G.D., Hesdorffer D., Logroscino G., Hauser W.A.Morbidity of nonfebrile status epilepticus in Rochester, Minnesota, 1965-1984 //
Epilepsia. - 1998. - Vol. 39/8. - P. 829-832.
9. Cockerel O.C., Walker S.M., Sander J.W., Shorvon S.D.Complex partial
status epilepticus: a recurrent problem // J. Neurol. Neurosurg Psychiatry. -
1994. - Vol. 57. - P. 835-837.
10. DeLorenzo R.J., Garnett L.K., Towne A.R. et al.Comparison of status epilepticus with prolonged seizure episodes lasting from 10 to 29 minutes //
Epilepsia. - 1999. - Vol. 40/2. - P. 164-169.
11. Philips S.A., Shanahan R.J.Etiology and mortality of status epilepticus in children // Arch Neurol. - 1989. - Vol. 46 - Р. 74-76.
12. Sander J.W.A.S., Hart Y.M., Trevisol-Bittencourt P.S.Absense status //
Neurology. - 1990. - Vol. 40. - P. 1010.
13. Shorvon S.D.Status epilepticus: its clinical features and treatment in children and adults. - Cambridge: Cambridge University Press, 1994.
14. Wasterlain C.G. and Treiman D.M.Status Epilepticus: mechanisms and management. - London: The MIT Press, 2006.
15. Treiman D.M.Status epilepticus // In: Textbook of epilepsy. - Eds. J. Laidlaw, A. Richens, D. Chadwick. - Edinburgh: Churchill-Livingstone,
1993. - P. 205-220.
Згідно з визначенням Міжнародної протиепілептичної лізі (International League Against Epilepsy - ILAE) 2005р., Епілептичний напад - тимчасовий клінічний прояв патологічної надлишкової або синхронної нейронної активності головного мозку. 
Для правильної діагностики епілепсії спочатку необхідно встановити тип епілептичного нападу відповідно до сучасної міжнародної класифікації епілептичних припадків, використовуючи нове визначення терміна «епілепсія».
Перший етап діагностики - це збір інформації про сам нападі, його феноменології, ймовірності його провокації; оптимально при наявності відео самого нападу.
Другий етап діагностики - після встановлення факту епілептичного нападу необхідно встановити його тип, згідно з класифікацією. У 1981 р .. Прийнята класифікація епілептичних припадків, але дискусії щодо її удосконалення тривають. З 2016 представлена оновлена робоча класифікація епілептичних припадків, яка може використовуватися в практиці, але буде остаточно прийнята пізніше, очікувано в 2017
Класифікація епілептичних нападів (ILAE, 2016), базова схема:
1. Фокальні:
- моторні;
- ходові;
- білатеральні тоніко-клонічні.
2. Генералізовані:
- моторні;
- абсанси.
3. З невідомих початком:
- моторні;
- ходові.
4. Некласифіковані.
Для всіх нападів необхідно вказати рівень порушень свідомості: припадок без порушення свідомості, з порушенням свідомості, з невідомої свідомістю.
Класифікація фокальних епілептичних нападів (ILAE, 2016):
1. Моторні:
- тонічні;
- атонические;
- міоклонічні;
- клонічні;
- епілептичні спазми;
- гіпермоторних.
2. ходові:
- сенсорні;
- когнітивні (галюцинації, дежавю, ілюзії, порушення уваги, афазія, нав'язливі думки)
емоційні (ажитація, агресія, плаксивість, сміх) - вегетативні (бради-, тахікардія, асистолія, відчуття холоду або жару, почервоніння або блідість шкіри, гастроінтестинальні порушення, лихоманка, гіпер-, гіповентиляція, нудота, блювота, пілоерекція і ін.)
- автоматизми (агресії, мануальні (в руках), орофаціальна, сексуальні, вокализация, складні рухи у вигляді ходьби або бігу, роздягання).
3. Білатеральні тоніко-клонічні (в минулому класифікації - з вторинною генералізацією).
Для фокальних нападів також для кожного типу нападу необхідно відзначити ступінь свідомості: напад без порушення свідомості, з порушенням свідомості, з невідомої свідомістю.
З 2016 ILAE внесла деякі зміни в термінологію нападів. Так, рекомендується замінити термін «часткові» нападу на «фокальні» (с / без порушення свідомості і з невідомої свідомістю), «комплексні парціальні» нападу - на «фокальні з порушеною свідомістю».
Майже 60% епілептичних припадків є локальними і тільки 23% - генералізованими тоніко-клонічними. Згідно з даними сучасних досліджень, епілепсія є системним захворюванням головного мозку, пов'язаних з порушенням нейрональних зв'язків, а не тільки локальним порушенням функції головного мозку. За залученню багатьох нейрональних зв'язків епілептичні припадки можуть виникати з неокортикальних, таламо-кортикальних, лимбических і стовбурових відділів.
Третій етап діагностики. Крім встановлення типу нападу, необхідно провести топічної діагностики нападу, тобто встановити місцезнаходження епілептичного вогнища, якщо ці напади є локальними. Напади, що виникають внаслідок надмірного патологічного збудження певної групи нейронів в різних частках головного мозку, мають свої характеристики.
Темпоральні епілептичні припадки: денні, виникають з частотою кілька разів на місяць, рідко ускладнюються епілептичним статусом, маніфестують ходові феноменами - часто аурою (вегетативної, психотической, часто з порушеннями свідомості, автоматизмами (оральними, вербальними), моторні феномени рідкі, з них можуть бути дистонические установки, постіктальном порушення свідомості.
Лобові епілептичні припадки мають наступні характеристики: часті кластерні атаки, короткочасний перебіг (20-40 с), часто розвиток уві сні, часто з вторинною генералізацією до епістатусного течії, поліморфні аури з різким початком, рано дебютують передомінантние моторні зміни - парези, паралічі, дисграфія і т.п., можуть протікати з порушенням свідомості, відновлення після нападу швидке. Найчастіше діагностують тонічні лобові нападу (близько 64%), потім клонічні (36%) і епілептичні спазми (36%).
Для фокальних епілептичних припадків з вогнищами в задній корі характерні зорові, соматосенсорні, вегетативні, смакові аури, адверсівних нападу і опсоклонуси очей, моргання, анозогнозия, акалькулия, апраксія, алексія.
Четвертий етап діагностики. Типи епілептичних припадків є основою для встановлення форми епілепсії, відповідно до класифікації тисяча дев'ятсот вісімдесят дев'ять Згідно з визначенням 2005р., Епілепсія - розлад головного мозку, що характеризується стійкою схильністю до епілептичних припадків, а також Нейробіологічні, конітівніми, психологічними і соціальними наслідками цього стану. Це визначення епілепсії передбачає розвиток як мінімум одного епілептичного нападу. Термін «розлад» недостатньо зрозумілий для пацієнтів і прізменшуе серйозність стану, тому ILAE і Міжнародне бюро по вивченню епілепсії (International Bureau for Epilepsy - IBE) недавно прийняли спільне рішення вважати епілепсію хворобою. У 2014 прийнято нове практичне визначення епілепсії, згідно з яким епілепсія - захворювання головного мозку, що відповідає наступним станам:
- не менше двох неспровокованих або рефлекторних епілептичних припадків з інтервалом не менше 24 год;
- один неспровокований (рефлекторний) епілептичний припадок і ймовірність повторних нападів, яка відповідає загальному ризику рецидиву (\u003e 60%) після двох неспровокованих епілептичних припадків в наступні 10 років
- діагноз епілептичного синдрому (наприклад синдром Веста).
Критерії «завершення» епілепсії включають досягнення певного віку у пацієнтів з формою епілепсії, залежить від віку, або відсутність епілептичних нападів протягом 10 років у пацієнтів, які не отримували протисудомних препаратів протягом більше 5 років. Робоча група працювала над терміном «лікування», який вказує на те, що ризик епілептичних припадків не вище, ніж у здорових людей, Однак у пацієнтів з епілепсією в анамнезі такий низький ризик ніколи не досягається. Термін «ремісія» недостатньо зрозумілий і не говорить про відсутність хвороби. Робоча група запропонувала термін «завершення» епілепсії, який свідчить, що епілепсії у пацієнта вже немає, проте не можна з упевненістю виключити появу нападів в майбутньому. Ризик рецидиву нападів залежить від форми епілепсії, віку, етіології, лікування та інших чинників. Наприклад, при ювенільної миоклонические епілепсії ризик повторних нападів залишається високим протягом десятиліть. Структурні ураження головного мозку, вроджені вади супроводжуються постійною схильністю до епілептичних припадків. При дослідженні 347 дітей з відсутністю нападів протягом не менше 5 років (без прийому протисудомних препаратів) пізні рецидиви зареєстровані у 6% дітей.
причини епілепсії
Більше половини дітей з епілепсією страждають ідіопатичною форми захворювання, при якій немає інших встановлених причин, ніж генетичні. У класифікації AT Berg і співавторів (2010) замість терміна «ідіопатична» запропоновано «генетична», тобто в результаті вже відомих і передбачуваних генів. Багато генів вже відомо (аутосомно-домінантна нічна лобова епілепсія і ін.).
Епілепсія, причина якої відома і вона не пов'язана з генетичними факторами, називається симптоматичної (структурної / метаболічної), за термінологією AT Berg і співавторів (2010). В цьому випадку епілепсія є вторинним результатом конкретних встановлених структурних або метаболічних захворювань:
- ушкодження речовини головного мозку внаслідок хронічної гіпоксії і асфіксії в пологах, пологова травма, субдуральна гематоми, перенесені вроджені TORCH-інфекції;
- метаболічні захворювання (порушення обміну амінокислот, вуглеводів та ін.), які супроводжуються поліорганної симптоматикою, крім епілепсії;
мітохондріальні хвороби; - вроджені вади розвитку головного мозку;
- хромосомні синдроми: синдроми Ангельмана, Дауна, ламкою Х-хромосоми і ін .;
- спадкові невротичні синдроми (Факоматози): склероз туберози і ін .;
- черепно-мозкова травма;
- судинні артеріовенозні мальформації головного мозку;
- перенесений інсульт.
Існує ще одна група епілепсії з невідомою етіологією (раніше називалася криптогенная епілепсія), причина нападів при якій поки не встановлена, вона може бути генетичної або структурно-метаболічної.
Прогноз залежить від обсягу і причини поразки мозку. Так, важкі пренатальні ураження можуть важко піддаватися лікуванню.
Міжнародна класифікація епілепсії та епілептичних синдромів ILAE 1989 (скорочений варіант)
Локалізаційно обумовлені (фокальні, парціальні) епілепсії і синдроми
1. Идиопатические (генетичні):
- доброякісна епілепсія дитячого віку з центрально-темпоральними спайками на електроенцефалограмі (ЕЕГ) (роландична)
- доброякісна дитяча епілепсія з потиличними нападами (синдром Гасто)
- доброякісна парціальна потилична епілепсія з раннім дебютом (синдром Панайотопулоса)
первинна епілепсія при читанні; - аутосомно-домінантна лобова епілепсія.
2. Симптоматичні (структурні / метаболічні):
- хронічна прогресуюча парціальна епілепсія дитячого віку (Кожевнікова)
синдром Расмуссена; - епілепсія, яка характеризується нападами, що викликаються специфічними провокуючими факторами;
- скронева епілепсія;
- лобова епілепсія;
- тім'яна епілепсія;
- потилична епілепсія.
3. Кріптогенние (невідомі).
1. Идиопатические (генетичні) епілепсії
Доброякісна дитяча епілепсія з центро-темпоральними спайками на ЕЕГ (роландична епілепсія)
Частота в популяції 21 на 100 тис. Дитячого населення.
Діагностують у 15-25% серед усіх дітей шкільного віку з епілепсією. Хвороба дебютує у віці 4-10 років з максимумом в 9 років. Хлопчики хворіють частіше, ніж дівчата. клінічно проявляється характерними ознаками: Початок з сенсомоторної аури, з'являються «горлові» звуки або анартрия, геміфаціальний короткі моторні напади вночі при засипанні і пробудження, в 20% - також фаціобрахіальні судоми, в 25% випадків в дебюті спостерігають вторинно генералізовані припадки. Тривалість нападів: прості - 30-60 с, вторинно генералізовані - до 1-2 хв з частотою нападів 2-6 разів на рік (у віці до 6 років в дебюті хвороби - часті напади). Ця форма є доброякісною, тобто крім епілептичних припадків немає змін в неврологічному статусі, когнітивної сфері - дитина може вчитися в середній загальноосвітній школі. Хвороба має доброякісний перебіг; ремісія настає зазвичай в 98% пацієнтів до досягнення пубертатного періоду.
Епілептиформні зміни між нападами в 90% випадків;
типово: доброякісні епілептиформні зміни дитинства (ДЕЗД) в центрально-скроневих відведеннях (по типу QRST на ЕКГ), але у віці 3-5 років - в задньоскронево-потиличних відведеннях;
у 30% дітей реєструють тільки нічні ЕЕГ-феномени (під час повільного сну - пік-хвильові комплекси) нормалізація ЕЕГ відбувається значно пізніше, ніж клінічна ремісія.
У лікуванні застосовують тільки монотерапії одним з препаратів першої лінії препарати вальпроєвої кислоти, карбамазепін, ламотриджин, окскарбазепін, габапентин, топірамат, леветірацетам. Але є дані про можливу вторинної білатеральної синхронізації, особливо при застосуванні карбамазепіну і окскарбазепіну.
Доброякісна парциальная потилична епілепсія з раннім дебютом (синдром Панайотопулоса)
Напади виникають рідко (до 5-7 за життя), переважно під час сну, проявляються девіацій очей в сторону, порушенням свідомості по типу дезорієнтації, активним блювотою, після цього виникає нападоподібний головний біль. У половини дітей напади можуть бути тривалими - протягом декількох годин з втратою свідомості (іктальне непритомність), супроводжуються блювотою, девіацій очей, клоническими гемісудомами, постіктальном головним болем.
Дитяча потилична епілепсія з пізнім дебютом (синдром Гасто)
Напади реєструють частіше, ніж при синдромі Панайотопулоса (1 раз в тиждень - 1 раз на місяць). Хвороба починається у віці 3-15 років, з максимумом в 8 років. Клінічне ядро - прості парціальні сенсорні нападу - зорові галюцинації в периферичному полі зору, геміаноптічні галюцинації, ілюзії з відчуттям болю в очах, моргання, поворот очей і голови в протилежну від епілептогенного вогнища сторону. Тривалість нападів становить секунди-хвилини. В кінці нападу характерні скарги на сильний головний біль з блювотою (у 50% хворих). Може бути вторинна генералізація з тоніко-клонічними судомами. При синдромах Панайотопулоса і Гасто змін при оцінці неврологічного статусу і когнітивної сфери дитини немає.
- ДЕЗД в потиличних відведеннях у 90% хворих між нападами;
- основний фон без змін;
- У 30% дітей можуть бути зміни в скроневих відведеннях;
- типово: зникнення патологічного патерну при відкриванні очей висока фотосенсітівність;
- нічний ЕЕГ-відеомоніторинг: в стадії повільного сну - наростання ДЕЗД-комплексів ( рання діагностика хвороби) нормалізація ЕЕГ-картини до досягнення віку 15 років.
У лікуванні застосовують принцип МОНОТЕРАПІЇ одним з наступних препаратів - карбамазепін, препарати вальпроєвої кислоти, окскарбазепін, топірамат, ламотриджин.
Зазначені форми епілепсії також вважають доброякісними. Повна ремісія при синдромі Панайотопулоса виникає до досягнення віку 9 років, при синдромі Гасто - 15 років.
Аутосомно-домінантна лобова епілепсія
Гени CHRNA4, CHRNA2, CHRNB2 локалізовані в локусах 20q13, 8q, 1p21 відповідно. Ця форма ідіопатичної епілепсії починається частіше у віці 7-12 років. Характерні нічні напади (після засипання, за 2-3 години до пробудження). Початок виникає з вокалізації (зазвичай - крик), при цьому очі відкриті. За характером нападу прості і складні парціальні.
Характерний поліморфізм клініки нападів - складні рухові акти: дитина сідає, чеше ніс, голову, робить гримаси, жувальні рухи, стає на карачки, розгойдується, робить педалювальні або боксувальні руху. У 70% випадків може бути аура (неприємні звуки, генералізований озноб, запаморочення) - дитина прокидається. Тривалість нападу - до 1 хв. За ніч може бути кілька нападів. При цій формі епілепсії існує тенденція до серійності і «світлий проміжок» (відсутність нападів протягом 2-3 міс). Обстеження не виявляє змін в неврологічному статусі, інтелекті й мови.
Характеристики ЕЕГ:
- основний фон - без змін;
- в стані поза сном - без епілептичних феноменів;
- основна діагностична методика - нічний ЕЕГ-відеомоніторинг, під час якого реєструють регіональну актівність в лобових, лобно-скроневих відведеннях.
Лікування складне, частіше ефективна политерапия: карбамазепін, препарати вальпроєвої кислоти, топірамат, ламотриджин, леветірацетам або комбінація базових препаратів.
Ця форма епілепсії вимагає проведення диференціального діагнозу з симптоматичною лобової епілепсію, при якій на ЕЕГ спостерігається уповільнення основного ритму, неврологічний статус - без вогнищевих змін, при нейровізуалізації - органічні зміни речовини мозку. Також слід провести диференційний діагноз з парасомніі, при яких на ЕЕГ відсутні епілептичні патерни.
2. Симптоматична (структурна / метаболічна) епілепсія
лобова епілепсія
Серед усіх симптоматичних і ймовірно симптоматичні (криптогенний) епілепсії симптоматична лобова епілепсія становить 20%. Вона може починатися в будь-якому віці в залежності від причини. Залежно від локалізації епілептогенного вогнища виділяють 7 форм лобової епілепсії, і кожна проявляється своїми типами нападів. В цілому характеризується локальними простими або складними нападами, які виникають в лобовій корі - контралатеральні клонічні судоми, одно-, двосторонні тонічні судоми, які закінчуються паралічем Тодда, складні автоматизми, які виглядають як молотильні руху кінцівками, розгойдування тулуба, педалювальні руху ногами. Епілептичні розряди в додаткової лобової моторної області проявляються складними фокальними нападами у вигляді тонічнихсудом рук, класичної «пози фехтувальника», адверсія голови, двостороннього розгинання тулуба, шиї, вокалізації. Активність в області повороту голови і очей проявляється адверсія очей в протилежну сторону, морганням. Свідомість збережена або втрачається в повному обсязі. Напади з фокусом в центральній зоні (ділянка кори у роландова борозни) характеризуються джексоновского маршем або строго локалізованими клоническими або тонічними судомами, судомами особи, втратою м'язового тонусу. При подразненні шкіри може виникнути моторний напад без порушення свідомості, фаціальні судоми з ковтальними, жувальними рухами, саливацией з почуттям іншого смаку, гортанні симптоми. Напади нічні, дуже часто, короткострокові.
У неврологічному статусі виявляють парези, атаксія, інтелектуальні і мовні порушення.
Характеристики ЕЕГ:
- основна фонова активність уповільнена;
- регіональна епіактівність (гострі хвилі, комплекси гостра - повільна хвиля, пік-хвилі)
біфронтальна або дифузна активність; - вторинна билатеральная синхронізація (ознака погіршення хвороби, поява когнітивних порушень).
Лікування складне. Дуже часто напади резистентні до адекватної терапії. Жінкам необхідно рекомендувати розпочинати з монотерапії препаратами першої лінії в адекватній дозі, а далі переходити на комбінацію препаратів з різними механізмами дії, згідно Однаковим протоколом лікування епілепсії у дітей 2014 Препарати першої лінії - карбамазепін (при вторинної білатеральної синхронізації протипоказаний), окскарбазепін, топірамат, другий - препарати вальпроєвоїкислоти, ламотриджин, третьої - комбінації препаратів.
скронева епілепсія
Часта форма всіх симптоматичних епілепсії (30-35%). Дебют відзначають в різному віці (частіше шкільному). часті причини: Наслідки гіпоксично-ішемічної енцефалопатії у вигляді глиозом, вроджені вади розвитку (кортикальна дисплазія), арахноїдальні кісти, наслідки перенесеного енцефаліту, формування склерозу гіпокампа. Напади можуть бути у одного пацієнта з / без порушення свідомості. Напади тривалі - 1-2 хв. Вегетативні прояви, психічні та сенсорні симптоми наявні протягом усього нападу або тільки на початку у вигляді аури, далі триває фокальний напад з порушенням свідомості з білатеральних тоніко-клонічними судомами. Виділяють дві форми скроневої епілепсії в залежності від епілептогенного вогнища: медійна (амігдалу-гіпокампальна) і латеральна (неокортикальних) епілепсія.
Медійна (амігдалу-гіпокампальна) епілепсія займає 65% всіх скроневих епілепсій і обумовлена наявністю фокусу в медіальних відділах скроневої частки. Причина - гіпокампальні атрофії, часто у пацієнтів, у яких у віці до 3 років спостерігалися комплексні фебрильні судоми, особливо тривалі односторонні атаки (в 40% випадків). Після 5-6-річного періоду ремісії починаються фокальні часті резистентні нападу, тобто розвивається хронічна епілепсія.
Клінічна основа цього підтипу епілепсії:
- фокальні напади без порушення свідомості - ізольована аура (вегетовісцеральних, нюхові і смакові галюцинації), психічні феномени - стан сну, деперсоналізації, дереалізації, страх, афект, радості, ороаліментарні автоматизми із збереженою свідомістю, дистонічного положення контралатеральної руки, в ипсилатерально руці можуть бути прості автоматизми;
- фокальні напади з ізольованим виключенням свідомості і автоматизмами без судом (діалептічні припадки).
Для латеральної (неокортикальної) епілепсії характерні:
- слухові галюцинації
- зорові яскраві галюцинації (панорамні види)
- вегетативні напади (несистемного запаморочення, «скроневі синкопе» - повільне падіння без судом з дистонічного установкою кінцівок, автоматизмами)
- пароксизмальна сенсорна афазія.
Крім частих нападів, при грубих осередкових змінах речовини мозку діти мають неврологічний дефіцит контралатерально вогнища (парез), емоційні і інтелектуальні порушення.
Характеристики ЕЕГ:
- у 50% хворих нормальна картина ЕЕГ між нападами;
необхідний стандарт дослідження - інвазивні електроди; - у 30% хворих спостерігаються епіпатерни між нападами;
- при медіальної епілепсії - зміни в передньоскроневіх відведеннях;
- ЕЕГ-осередки можуть не збігатися з морфологічним вогнищем на магнітно-резонансної томографії (МРТ) - формування «дзеркального» осередку;
- характерний ЕЕГ-феномен в дебюті - продовжено регіональне уповільнення активності;
- провокація - іноді депривація сну;
- нічна ЕЕГ свідчить про 65% змін між нападами.
На інтеріктальном ЕЕГ - передній скронева фокус спайок, пароксизмальної тета-ритм.
Характеристики змін на МРТ головного мозку при медіальної епілепсії - атрофія гіпокампа, посилений сигнал на Т2 від гіпокампу. Склероз гіпокампа прогресує.
Лікування хірургічне. Прогноз після хірургічного лікування хороший. Медикаментозне лікування складне і не завжди ефективне; часто застосовують політерапію.
тім'яна епілепсія
Напади суб'єктивні, тому виявити їх складно, особливо у дітей молодшого віку. Характерні соматосенсорні нападу у вигляді чутливого джексоновского маршу, часто пов'язані з моторними феноменами. Соматосенсорні симптоми можуть бути позитивними і негативними, можливі болі в животі, нудота, ілюзія руху, відсутність почуття тіла (асоматогнозія), запаморочення, дезорієнтація в просторі. Порушення сприйняття й мови (із залученням домінантної півкулі), постуральні або ротаторні руху, при поширенні імпульсу можуть розвиватися зорові симптоми (потилично-скронево-тім'яна), контралатеральні або ипсилатерально руху з дистонічного становищем кінцівки в протилежну сторону або в сторону залученої півкулі. Зорові ілюзії (макропсия, микропсия, метаморфопсії) свідчать про наявність розрядів в задніх відділах тім'яної кори і паріетотемпороскроневоі частки.
потилична епілепсія
Реєструють у 5% дітей всіх симптоматичних і криптогенним форм.
Епілептичні розряди по первинної зорової кори виявляються:
- окоруховими розладами (ністагм, девіація очей в протилежну сторону, двосторонній міоз)
- локальними нападами без порушення свідомості у вигляді зорових галюцинацій, ілюзій, пароксизмальної амавроза, звуження полів зору;
- локальними нападами з порушенням свідомості і з білатеральних тоніко-клонічними нападами;
- вегетативні розлади кінці нападу (головний біль, блювота)
- акалькулия, апраксія.
- Неврологічний дефіцит залежить від причини епілепсії. Часто виявляють окорухових порушення (порушення конвергенції, косоокість).
Характеристики ЕЕГ:
- між нападами може бути в нормі;
- уповільнення основного фону;
- одностороннє придушення альфа-ритму при грубих органічних змінах;
- ЕЕГ-патерни не змінюються при відкриванні очей (диференційний діагноз з ідіопатичною потиличної епілепсію)
- поширення епіактівності на скроневі відведення;
- провокація - фотостимуляція.
Злоякісні мігруючі фокальні напади раннього дитинства (синдром Коппола - Дюлак)
Відносно нова форма фокальній епілепсії.
характеристика:
- етіологія невідома (імовірно генетичного походження);
- початок вік 6 міс;
- нормальний розвиток до дебюту;
- моторний і інтелектуальний регрес;
напади:
- фокальні моторні;
- білатеральні тоніко-клонічні;
- вегетативні (апное, ціаноз)
- у вигляді серій і кластерів (2-5 доби), короткі ремісії.
прогресуюча мікроцефалія;
на ЕЕГ - типовий фокальний патерн в різних відведеннях;
на МРТ - норма.
Лікування: препарати першої лінії - топирамат, ламотриджин, другий - препарати вальпроєвої кислоти, леветірацетам.
висновки
Використання нового визначення епілепсії і нової класифікації нападів дозволяє враховувати більшість типів епілептичних припадків і привести термін «епілепсія» у відповідність з термінологією, яка використовується більшістю лікарів, які займаються питаннями епілепсії.
В останні роки синтезовано багато нових протиепілептичних препаратів для поліпшення якості лікування пацієнтів: в період 2007-2012 рр. - еслікарбазепіну ацетат (eslicarbazepine acetate), лакосамід (lacosamide), перампанел (perampanel), ретігабін (retigabin), руфінамід (rufinamid), стіріпентол (stiripentol), в 2016 - бріварацетам (brivaracetam). Але в період дитинства золотим стандартом залишаються протиепілептичні препарати з широким спектром дії - препарати вальпроєвої кислоти, ламотриджин, топірамат, карбамазепін, які є базовими.
Епілептичні синдроми у дітей: діагностика та лікування
Мухін Костянтин Юрійович
Петрухін Андрій Сергійович
епілепсія- одна з найбільш актуальних проблем в педіатричній неврології. Частота епілепсії в дитячій популяції становить 0,5-0,75% дитячого населення, а фебрильних судом - до 5%. З жалем слід констатувати, що в нашій країні до теперішнього часу існує велика плутанина у встановленні діагнозу епілепсії, що значно спотворює статистику. Такі діагнози, як "епілептиформні синдром", "судомний синдром", "підвищена судомна готовність" та ін., По суті, є на епілепсію і повинні бути чітко систематизовані відповідно до Міжнародної класифікації. Мета цього видання - ознайомити лікарів з класифікацією, принципами діагностики і лікування основних форм епілепсії у дітей.
Визначення. Епілепсія є хронічне захворювання головного мозку, що характеризується повторними непровоціруемимі нападами порушень рухових, чутливих, вегетативних, розумових або психічних функцій, що виникають внаслідок надмірних нейронних розрядів. Представлене визначення містить два важливих положення. По-перше, епілепсія не включає поодинокі напади незалежно від їх клінічних проявів. Тільки повторні напади є підставою для встановлення діагнозу епілепсії. По-друге, до епілепсії відносяться спонтанні, непровоціруемие напади (виняток - рефлекторні форми). За визначенням, фебрильні судоми, а також судоми, що виникають при гострих захворюваннях головного мозку (наприклад, при енцефаліті, субдуральної гематоми, гострому порушенні мозкового кровообігу та ін.), Не є на епілепсію.
Класифікація. Прояви епілепсії вкрай різноманітні, що вже з самого початку вивчення захворювання ускладнювало створення єдиної класифікації. Сучасна класифікація епілептичних нападів була прийнята Міжнародною Лігою по боротьбі з епілепсією в 1981 році в місті Кіото (Японія). На відміну від попередніх класифікацій, вона враховує як клінічні, так і нейрофізіологічні (ЕЕГ) критерії більшості видів епілептичних нападів (табл. 1). Класифікація поділяє всі види епілептичних нападів на парціальні (вогнищеві, фокальні, локальні, локалізаціонно-обумовлені), генералізовані і неклассіфіціруемие. Парціальні напади діагностуються в тому випадку, коли на початку пароксизму є чіткі клінічні та електрофізіологічні критерії залучення певних структур головного мозку. У тому випадку якщо напад починається як парціальний, а потім відбувається залучення всієї мускулатури тулуба і кінцівок і ознаки залучення обох гемисфер на ЕЕГ, то його слід класифікувати як вогнищевий з вторинною генералізацією.
У класифікації уточнено поняття простих і складних парціальних нападів. Прості парціальні - напади без виключення свідомості. Під складними парціальними нападами слід розуміти пароксизми з повним виключенням свідомості. За сучасною класифікацією всі напади, що протікають з явищами деперсоналізації, сновідних станами, когнітивними розладами та ін., Відносяться не до складних, а до простих парціальним, так як свідомість пацієнта під час даних пароксизмів змінено, але не виключене і зберігається пам'ять про приступи. Також класифікація 1981 року передбачає, що у одного пацієнта може бути декілька
різних типів нападів. Наприклад, напад, розпочавшись як простий парціальний, може трансформуватися в складний парціальний, а потім у вторинно-генералізований. З класифікації вилучено термін "поліморфні напади", який не несе будь-якої інформації і не рекомендується до вживання. Таким чином, класифікація Кіото є на даному етапі найбільш повною систематизацією епілептичних нападів.
З накопиченням клінічного досвіду, впровадженням в практику методу відео - ЕЕГ-моніторингу, розвитком методів нейровізуалізації, молекулярної генетики та інших наук, стало очевидним, що існує цілий ряд особливих форм епілепсії, для яких характерна своя клініка (типові види нападів), перебіг і прогноз . Деякі з цих форм були відомі давно, як, наприклад, синдром Веста, синдром Леннокса-Гасто, роландична епілепсія. Інші -доброякісні сімейні неонатальні судоми, важка миоклоническая епілепсія дитинства та ін. - виділені лише в останні роки. Дані форми епілепсії, або, за Міжнародною класифікацією, епілептичні синдроми, як правило, проявляються не яким-небудь одним видом нападів, а їх поєднанням. Епілептичні синдроми визначаються як окремі, самостійні форми епілепсії, які характеризуються лімітованим віком дебюту нападів, наявністю особливого виду нападів, специфічних змін на ЕЕГ (характерних для даного синдрому), закономірностями перебігу і прогнозу. Наприклад, один вид нападів - абсанси - може входити в структуру цілого ряду епілептичних синдромів: дитяча та юнацька абсанс епілепсія, юнацька міоклонічна епілепсія, епілепсія з миоклонические абсансах і інші, причому особливості перебігу та прогноз при всіх зазначених синдромах різні.
Принципово новим кроком у розвитку епілептології було створення сучасної класифікації "епілепсії, епілептичних синдромів і асоційованих з нападами захворювань". Дана класифікація була прийнята Міжнародною Лігою по боротьбі з епілепсією в жовтні 1989 року в Нью-Делі і в даний час є загальноприйнятою для епілептології усього світу (табл. 2).
Класифікація епілептичних синдромів базується на наступних принципах:
Принцип локалізації:
локалізаціонно-обумовлені (фокальні, локальні, парціальні) форми епілепсії;
генералізовані форми;
форми, що мають риси як парціальних, так і генералізованих.
Принцип етіології:
симптоматичні,
криптогенні,
идиопатические.
Вік дебюту нападів:
форми новонароджених,
дитячі,
дитячі,
юнацькі.
Основний вид нападів, що визначає клінічну картину синдрому:
абсанси,
міоклонічні абсанси,
інфантильні спазми і ін.
Особливості перебігу і прогнозу:
доброякісні;
важкі (злоякісні).
Класифікація поділяє всі форми епілепсії на симптоматичні, ідіопатичні і Криптогенні. Під симптоматичними формами маються на увазі епілептичні синдроми з відомою етіологією і верифікованими морфологічними порушеннями (пухлини, рубці, глиоз, кісти, дізгенезіі і ін.). При ідіопатичних формах відсутні захворювання, що можуть бути причиною епілепсії, і епілепсія є як би самостійним захворюванням. В даний час встановлена генетична детермінованість идиопатических форм епілепсії. Термін "криптогенний" (прихований) відноситься до тих синдромам, причина яких залишається прихованою, неясною. Дані синдроми не задовольняють критеріям идиопатических форм, але немає доказів і їх симптоматичного характеру. Наприклад, в разі поєднання епілепсії з гемипарезом або олігофренією передбачається симптоматичний характер захворювання, але при КТ і МРТ дослідженні зміни в мозку не візуалізується. Даний випадок класифікується як криптогенний. Очевидно, що при вдосконаленні технічних можливостей нейровізуалізації (наприклад, ПЕТ), більшість кріптогенних форм буде переведено в розряд симптоматичних.
Симптоматика. Семіологія і клініка різних епілептичних нападів детально описана в багатьох посібниках з епілепсії. Разом з тим, діагностиці окремих форм епілепсії (епілептичних синдромів), в літературі приділено недостатньо уваги. Зупинимося на умовах діагнозу і принципах терапії основних епілептичних синдромів дитячого і підліткового віку.
Дитяча абсанс епілепсія
абсанси - різновид генералізованих бессудорожних нападів, що характеризується високою частотою і короткою тривалістю пароксизмів з виключенням свідомості і наявністю на ЕЕГ специфічного патерну - генералізованої пік-хвильової активності з частотою 3 Гц.
Дебют абсансов при дитячій абсанс епілепсії (ДАЕ) спостерігається у віковому інтервалі від 2 до 9 років, складаючи в середньому 5,3 років. Віковий пік маніфестації - 4-6 років, з переважанням по підлозі дівчаток.
Клінічно абсанси характеризуються раптовим коротким виключенням (або значним зниженням рівня) свідомості з відсутністю або мінімальними моторними феноменами. Аура, як і постпріступном сплутаність, не характерні. Тривалість абсансов коливається від 2-3 до 30 секунд, складаючи в середньому 5-15 сек. Характерною особливістю абсансов є їх висока частота, що досягає десятків і сотень нападів на добу.
Принциповим є поділ абсансов на прості і складні. Прості абсанси характеризуються припиненням будь-якої діяльності, "застигання", "завмиранням" пацієнтів, фіксованим "відсутнім" поглядом, розгубленим гипомимично виразом обличчя. Прості абсанси в клінічній картині ДАЕ складають лише 20%. Для ДАЕ більш характерні складні абсанси, що протікають з мінімальним моторним компонентом. Розрізняють такі види складних абсансов: з міоклоніческім, тонічним, атоническим, вегетативним компонентом, а також з автоматизмами і фокальними феноменами. Найбільш часто констатуються абсанси з міоклоніческім і тонічним компонентом.
Абсанси з міоклоніческім компонентом констатуються у 40% хворих ДАЕ. Виявляються: міоклонію століття; періоральний міоклонусом (ритмічне витягування губ на зразок "золотої рибки"); періназальним міоклонусом (ритмічне посмикування крил носа). У ряду хворих під час нападу відзначається одноразове здригання або кілька коротких слабких посмикувань м'язів плечового пояса і / або рук.
Абсанси з тонічним компонентом найбільш типові для ДАЕ, спостерігаючи у 50% хворих. Виявляються відхиленням голови, а іноді і тулуба, тому (ретро-пульсівние абсанси), тонічним відведенням очних яблук вгору або в сторону. Іноді під час нападу спостерігається легке тонічне напруга (частіше асиметричне) м'язів верхніх кінцівок.
Абсанси з атоническим компонентом не характерні для ДАЕ і констатуються лише в одиничних випадках, в основному при атипових формах. Виявляються раптовою втратою м'язового тонусу в м'язах рук (випадання предметів), шиї (пасивний кивок), ніг (атонически-астатические напади). Атонічні абсанси частіше ставляться до атиповим Абсанс (частота пік-хвильових комплексів менше 2,5 Гц) в рамках синдрому Леннокса-Гасто.
Абсанси з вегетативним компонентом спостерігаються, в середньому, у 5% хворих ДАЕ. Виявляються нетриманням сечі в момент нападу, мідріазом, зміною кольору шкірних покривів обличчя і шиї з появою уртикарії на шкірі даних областей.
Абсанси з фокальним компонентом констатуються у 15% хворих і значно переважають у пацієнтів з тонічними абсансами. Під час нападу з'являється легке унилатеральное напруга м'язів руки або особи, іноді з одиничними миоклоническими посмикуваннями; поворот голови і очей в сторону. Слід пам'ятати, що поява виражених фокальних феноменів під час нападів насторожує щодо наявності у хворих парціальних нападів. Автоматизми в структурі абсансов спостерігаються у 35% пацієнтів, які страждають ДАЕ. Найбільш часто виникають автоматизми жестів, фарингіт-оральні і мовні.
Статус абсансов при ДАЕ відзначається з частотою близько 10%. Даний стан проявляється різким почастішанням абсансов, наступних один за іншим безпосередньо або з дуже коротким інтервалом. Спостерігається амимия, слинотеча, рухова загальмованість (ступор). Тривалість статусу становить від декількох годин до декількох діб.
Генералізовані судомні напади (ГСП) констатуються у 1/3 хворих ДАЕ. З моменту дебюту абсансов до приєднання ГСП проходить кілька місяців або років. У більшості випадків ГСП приєднуються через 1-3 роки після початку захворювання (65% в групі хворих з ГСП), рідше - в інтервалі 4-13 років (35%). Переважають рідкісні генералізовані тоніко-клонічні судомні пароксизми.
До факторів, що провокує почастішання абсансов, відносяться наступні: гіпервентиляція; депривація сну; фотостимуляція; напружена розумова діяльність або, навпаки, розслаблений, пасивний стан. Гіпервентиляція - основний провокуючий фактор виникнення абсансов. Проведення 3-хвилинної гіпервентиляції у нелікованих хворих ДАЕ викликає поява абсансов практично в 100% випадків; а у пацієнтів, які отримують АЕП, служить одним з критеріїв ефективності медикаментозної терапії.
Частота виявляється епілептиформні активності в міжнападу при ДАЕ висока і становить 75-85%. Найбільш типовий ЕЕГ патерн - спалахи генералізованої пік-хвильової активності. Частота пік-хвильових комплексів варіює від 2,5 до 4-5 в сек. (Зазвичай 3 Гц - типові абсанси).
Лікування. Повна терапевтична ремісія при ДАЕ досягається в 70-80% випадків і значне уражень нападів - у інших пацієнтів. Лікування завжди слід починати з препаратів вальпроєвої кислоти. Середні дозування складають 30-50 мг / кг / сут. в 3 прийоми. Показана також висока ефективність суксіміда в купировании абсансов. Середня дозування препарату становить 15 мг / кг / сут. в 2-3 прийоми. Істотним негативним моментом терапії суксімідом є повна відсутність впливу препарату на ГСП. При резистентності абсансов до монотерапії вальпроатами і сукцініміди призначається комбінація вальпроатов і суксіміда, або вальпроатов і ламотриджину (Ламіктал). Середня добова доза Ламікталу при поєднанні з вальпроатом становить 1-5 мг / кг / сут. в 2 прийоми. Застосування карбамазепіну (фінлепсин, тегретол, Тімон) категорично протипоказано при всіх формах абсансной епілепсії через високу ймовірність почастішання нападів.
Юнацька абсанс епілепсія
Юнацька абсанс епілепсія (ЮАЕ) - різновид ідіопатичною генералізованою епілепсії, що характеризується основним видом нападів - абсансами, дебютує в пубертатному періоді з високою ймовірністю приєднання ГСП і характерними ЕЕГ змінами у вигляді генералізованої пік-хвильової активності з частотою 3 Гц і більше.
Дебют абсансов при ЮАЕ варіює від 9 до 21 років, в середньому 12,5 років. У значної більшості хворих (75%) абсанси починаються в порівняно короткому часовому проміжку - 9-13 років. Важливою особливістю ЮАЗ є частий дебют захворювання з ДСП - 40% випадків.
Абсанси у пацієнтів, які страждають ЮАЕ, проявляються коротким виключенням свідомості з завмиранням і гіпоміміей. Характерно переважання простих абсансов, тобто нападів без будь-якого рухового компонента. Тривалість нападів становить від 2 до 30 сек., В середньому 5-7 сек. Разом з тим, у половини пацієнтів відзначаються дуже короткі абсанси, які не перевищують 3 сек. Характерною особливістю ЮАЕ є і відносно невисока частота нападів в порівнянні з ДАЕ. У більшості хворих переважають поодинокі абсанси протягом дня або 1 напад в 2-3 дня.
Генералізовані судомні напади констатуються у більшості хворих -75%. У групі хворих з ГСП захворювання частіше дебютує ні з абсансов, а з тоніко-клонічних судомних нападів. ГСП характеризуються короткими нечастими тоніко-клонічними судомами, що виникають зазвичай при пробудженні або засипанні.
На відміну від ДАЕ, гіпервентиляція провокує виникнення абсансов не більше ніж у 10% хворих ЮАЕ. ГСП у 20% хворих провокуються депривація сну.
При ЗЗГ дослідженні в міжнападу результати, близькі до норми, констатуються у 25% пацієнтів. Основним ЕЕГ патерном є генералізована пік-хвильова активність з частотою 3 Гц і більше (4-5 в сек.), Що носить переважно симетричний і білатерально-синхронний характер.
Лікування. Ефективність лікування при ЮАЕ достовірно нижче, ніж при ДАЕ. Терапевтична ремісія досягається в середньому у 60% хворих, значне уражень нападів - 35%, відсутність ефекту - 5%. Лікування починається з монотерапії препаратами вальпроєвої кислоти. Середня дозування становить 30-50 мг / кг / сут. З огляду на вкрай високу ймовірність при ЮАЕ приєднання ГСП, починати лікування з сукцінімідов, а також застосовувати їх у вигляді монотерапії не показано. При відсутності суттєвого ефекту від монотерапії вальпроатами в досить високих дозах, застосовується комбінація вальпроатов з сукцініміди або Ламікталом. Середні дози суксіміда складають 20 мг / кг / сут .; Ламікталу - 1-5 мг / кг / сут.
Епілепсія з ізольованими генералізованими судорожними нападами
Епілепсія з ізольованими ГСП визначається як синдром ідіопатичної генералізованої епілепсії, виявляється єдиним видом нападів - первинно-генералізованими тоніко-клонічними судорожними пароксизмами при відсутності аури і чіткого фокусу на ЕЕГ.
Дебют захворювання спостерігається в дуже широкому віковому діапазоні: від 1 до 30 років з максимум в пубертатному періоді (середній -13,5 років).
Клінічно ГСП проявляються раптовим (без аури) виключенням свідомості з падінням пацієнтів, судомами, закладом очних яблук, розширенням зіниць. Спочатку настає коротка тонічна фаза, що переходить в більш тривалу клонічними з подальшим постпріступном оглушением. Тривалість ГСП складає від 30 сек до 10 хв (в середньому 3 хв.). Частота нападів невисока - від одиничних в рік до 1 разу на місяць, без тенденції до серійного і статусному течією. Характерна приуроченість більшості нападів на період пробудження і, рідше, засипання. Найбільш значимий провокуючий фактор - депривація сну і раптове насильницьке пробудження. Можливо почастішання нападів в періменструальном періоді.
ЕЕГ дослідження в міжнападу у половини хворих (!) Може бути в межах норми. Характерна генералізована пік-хвильова активність з частотою від 3 Гц і вище, нерідко з амплітудною асиметрією або біфронтальним переважанням. Можуть виявлятися різні регіональні патерни.
Лікування. Ремісія досягається у 75-80% хворих. Базові препарати - карбамазепін і вальпроати. При відсутності на ЕЕГ генералізованої пік-хвильової активності лікування починається з карбамазепіну, який більш ефективний, ніж вальпроати. При приєднанні до генералізованим судомних нападів абсансов або міоклонічних нападів, або при появі на ЕЕГ генералізованої пік-хвильової активності базовий препарат - тільки вальпроати. Середня дозування карбамазепіну -15-25 мг / кг / сут. в 3 прийоми; вальпроатов 20-50 мг / кг / сут. в 3 прийоми. До резервних препаратів належать барбітурати (фенобарбітал 1.5-3.0 мг / кг / сут. В 1-2 прийоми; гексамидин, бензонал), гідантоїни (дифенін 4-8 мг / кг / сут. В 2 прийоми). У рідкісних резистентних випадках можливі комбінації: карбамазепін + вальпроати; карбамазепін + ламотриджин, карбамазепін + барбітурати; вальпроати + барбітурати. При неадекватному лікуванні можливе приєднання до ГСП абсансов або міоклонічних нападів з трансформацією в юнацьку миоклоническую епілепсію.
Юнацька міоклонічна епілепсія
Юнацька міоклонічна епілепсія (ЮМЕ) - одна з форм ідіопатичною генералізованою епілепсії, що характеризується дебютом в підлітковому віці з виникненням масивних білатеральних миоклонических нападів, переважно в руках, в період після пробудження пацієнтів. ЮМЕ - одна з перших форм епілепсії з відомим генетичним дефектом. Передбачається двулокусная модель успадкування (домінантно-рецесивна), причому домінантний ген локалізований на короткому плечі хромосоми 6.
Дебют ЮМЕ варіює від 7 до 21 років з максимум у віковому інтервалі 11-15 років. Захворювання може початися в більш ранньому віці з абсансов або ДСП, з подальшим приєднанням міоклонічних нападів в пубертатному періоді. Міоклонічні напади характеризуються блискавичними посмикуваннями різних груп м'язів; вони частіше двосторонні, симетричні, поодинокі або множинні, мінливі за амплітудою. Локалізуються головним чином в плечовому поясі і руках, переважно в розгинальних групах м'язів. Під час нападів хворі кидають предмети з рук або відкидають їх далеко в сторону. У 40% пацієнтів миоклонические напади захоплюють і м'язи ніг, при цьому хворий відчуває як би раптовий удар під коліна і злегка присідає або падає (міоклонічно-астатические напади); потім тут же встає. Свідомість під час нападів зазвичай збережено. Міоклонічні напади виникають або частішають в ранкові години, після пробудження пацієнта. У 90% випадків вони поєднуються з ГСП пробудження і в 40% - з абсансами. Основними провокуючими напади факторами є депривація сну і раптове насильницьке пробудження. Приблизно 1/3 хворих ЮМЕ (частіше жіночої статі) виявляють фотосенсітівная.
Епілептиформна активність на ЕЕГ виявляється у 85% хворих в міжнападу. Найбільш типова генералізована швидка (від 4 Гц і вище) полипик-хвильова активність у вигляді коротких спалахів. Можливо також поява пік-хвильової активності 3 Гц.
Лікування. Поряд з медикаментозною терапією необхідно строго дотримуватися дотримання режиму сну і неспання; уникати недосипання і чинників фотостимуляції в побуті. Базові препарати-виключно похідні вальпроєвої кислоти. Середня добова доза -40-60 мг / кг При недостатній ефективності призначається политерапия: вальпроати + суксилеп (при резистентних абсансах); вальпроати + фенобарбітал або гексамидин (при резистентних ГСП); вальпроати + ламотриджин або клоназепам (при резистентних миоклонических нападах і вираженою фотосенсітівності).
Повна медикаментозна ремісія досягається у 75% хворих, причому в більшості випадків на монотерапії вальпроатами. Однак в подальшому при скасуванні АЕП рецидиви констатуються у половини хворих. При цій формі епілепсії рекомендується відміна АЕП через не менш ніж 4 років з моменту настання ремісії.
Доброякісна дитяча парціальна епілепсія з центрально-скроневими піками (роландична епілепсія).
Роландична епілепсія (РЕ) - ідіопатична парціальна епілепсія дитячого віку, що характеризується переважно короткими геміфаціальний моторними нічними нападами, часто з попередньої соматосенсорної аурою і типовими змінами ЕЕГ.
Дебют РЗ варіює в віковому інтервалі від 2 до 14 років. У 85% випадків напади починаються у віці 4-10 років з максимумом в 9 років.
Спостерігаються прості парціальні (моторні, сенсорні, вегетативні), складні парціальні (моторні) і вторинно генералізовані судомні напади. Найбільш типові прості парціальні моторні та / або сенсорні пароксизми. Характерно початок нападу з соматосенсорної аури: відчуття поколювання, оніміння з одного боку в області глотки, мови, ясна. Потім з'являються моторні феномени: односторонні тонічні, клонічні або тоніко-клонічні судоми м'язів обличчя, губи, язика, глотки, гортані; фарингіт-оральні напади, часто поєднуються з анартрія і гіперсалівацією. При цьому уві сні хворі видають своєрідні горлові звуки типу "булькання", "хрюкання", "полоскання горла". У 20% пацієнтів судоми можуть поширюватися з м'язів обличчя на гомолатеральнимі руку (брахіофаціальние напади) і приблизно в 8% випадків втягують ногу. У міру розвитку захворювання напади можуть змінювати сторонность. Вдруге-генералізовані судомні напади відзначаються у 20-25% хворих РЕ. Тривалість нападів при РЕ невелика: від кількох секунд до 2-3 хв. Частота зазвичай невисока - в середньому 2-4 рази на рік. У перші місяці з моменту дебюту захворювання напади можуть бути частішими, проте з плином часу вони виникають все рідше, навіть без лікування. Пароксизми РЕ "жорстко" пов'язані з ритмом сон-неспання. Найбільш типові нічні напади, що виникають переважно при засипанні і пробудженні. Лише у 15-20% хворих напади спостерігаються як уві сні, так і в стані неспання.
На ЕЕГ в межприступном періоді з високою частотою виявляються характерні "роландіческой" пік-хвильові комплекси при обов'язково збереженій основний активності. Ці комплекси є повільні діфазние високоамплітудні піки або гострі хвилі (150 - 300 мкВ), нерідко з подальшими повільними хвилями, загальною тривалістю близько 30 мс. Дані комплекси нагадують зубці ORS ЕКГ. Роландіческой комплекси зазвичай локалізовані в центральній і скроневої області; можуть спостерігатися какунілатерально (зазвичай контрлатерально геміфаціальний нападів), так і білатерально незалежно. Типовою є нестійкість ЕЕГ патернів, їх варіабельність від одного запису до іншого.
Лікування. Базовим препаратом є вальпроати. Середня дозування -20-40 мг / кг / сут. в 3 прийоми. При неефективності - перехід на карбамазепін -10-20 мг / кг / сут. в 2-3 прийоми. Політерапія неприпустима!
Повна терапевтична ремісія досягається практично в 100% випадків. Після 14-річного віку напади зникають (при лікуванні або спонтанно) у 93% хворих, а після 16 років - у 98%. З огляду на абсолютно сприятливий прогноз, деякі автори пропонують не призначати лікування за наявності точного діагнозу РЕ. Дана точка зору дискусійна.
Ідіопатична парціальна епілепсія з потиличними пароксизмами
Ідіопатична парціальна епілепсія з потиличними пароксизмами (доброякісна потилична епілепсія (ДЗЕ)) - форма ідіопатичної локалізаціонно-обумовленої епілепсії дитячого віку, що характеризується простими парціальними нападами із зоровими розладами і наявністю на ЕЕГ специфічної пік-хвильової активності в потиличних відведеннях.
Захворювання починається у віці 2-12 років з двома піками дебюту - близько 3 і 9 років. Типові прості парціальні сенсорні пароксизми із зоровими розладами. Характерні прості зорові галюцинації, фотопсии, зорові ілюзії (макро, мікропсіі). Можлива поява минущого амавроза і гомонимной квадрантной ге-міанопсіі. Під час нападу нерідко спостерігається версівний компонент з насильницьким поворотом очей і голови. При вогнищі в потиличній частці часто спостерігається іррадіація збудження наперед (в скроневу і лобову частку) з появою складних структурних галюцинацій; виключенням свідомості і виникненням вторинно генералізованих судомних нападів. Типово виникнення нападів уві сні, особливо при пробудженні пацієнтів. Напади нерідко супроводжуються мігренознимі симптомами: головним болем і блювотою. Перші напади у дітей раннього віку найбільш важкі і тривалі (до декількох годин і навіть діб).
На ЕЕГ визначається поява високоамплітудними пік-хвильової активності в одному з потиличних відведенні або біокціпітально незалежно. Морфологія патернів нагадує таку при роландической епілепсії. Характерно зникнення епіактівності при записі ЕЗГ з відкритими очима.
Лікування. Препарати вибору - похідні карбамазепіну. Середня добова доза-близько 20 мг / кг. При неефективності застосовуються вальпроати - 30-50 мг / кг / сут. Резервний препарат фенітоїн - 3-7 мг / кг / сут. Лікування здійснюється тільки монотерапией. Резистентні випадки необхідно диференціювати з симптоматичною потиличної епілепсію. Повна терапевтична ремісія відзначається в 95% випадків. Разом з тим, напади купіруються складніше і дози АЕП вище, ніж при роландической епілепсії.
синдром Веста
Синдром Веста (СВ) - резистентна форма генералізованої епілепсії раннього дитячого віку, що характеризується нападами у вигляді інфантильних спазмів, затримкою психомоторного розвитку і специфічними змінами ЗЗГ (гіпсрітмія). Віковий пік маніфестації - 4-7 міс. Виділяють симптоматичну (пороки розвитку мозку, склероз туберози, перинатальна енцефалопатія, тезаурісмози і т.д.) і кріптогенную форми. При симптоматичної формі психомоторне розвиток дітей страждає зазвичай з народження, при криптогенной - з моменту початку нападів.
Інфантильні спазми проявляються раптовим скороченням м'язів шиї, тулуба, кінцівок, які, як правило, білатеральні і симетричні. Найбільш типові флексорние спазми ( "салаамови напади") зі згинанням шиї, тулуба, рук; згинанням, приведенням і підведення ніг. Напади короткі, групуються в серії; частіше виникають відразу після пробудження пацієнтів.
У неврологічному статусі виявляється груба затримка психічного і моторного розвитку, нерідко тетрапірамідная симптоматика.
На ЕЕГ констатується гіпсарітмія - високоамплітудні нерегулярні, слабо синхронізовані аритмічний повільні хвилі з розрядами спайки.
лікування починається з вальпроатов, вігабатрин (Сабріл) або АКТГ. При кріптогенних випадках СВ застосовується АКТГ (або Синактен-депо) в дозі 0,1 мг / кг / сут. або преднізолон - 2-5 мг / кг / сут. При симптоматичному СВ-вальпроати (50-100 мг / кг / сут. І вище) як монотерапія або в комбінації з АКТГ. В останні роки, на думку багатьох авторів, рекомендується застосування вігабатрин в дозі 100 мг / кг / сут. Вігабатрін є препаратом абсолютного вибору в лікуванні СВ, обумовленого туберозний склерозом. При відсутності ефекту від монотерапії базові АЕП комбінуються з ламотриджином, карбамазепіном або бензодіазепінами.
Прогноз захворювання важкий, як щодо нападів, так і психомоторного розвитку. Більшість дітей є інвалідами, не здатними до самостійного життя. Збігом часу СВ трансформується в СЛГ (1/3 випадків) або мультифокальну епілепсію.
Синдром Леннокса-Гасто
Синдром Леннокса-Гасто (СЛГ) - епілептична енцефалопатія дитячого віку, що характеризується поліморфізмом нападів, когнітивними порушеннями, специфічними змінами ЕЕГ і резистентністю до терапії. Частота СЛГ становить близько 5% серед усіх форм епілепсії у дітей та підлітків; хворіють частіше хлопчики.
Захворювання дебютує переважно у віці 2-8 років (частіше 4-6 років). Якщо СЛГ розвивається при трансформації з синдрому Веста, то можливо 2 варіанти:
1. інфантильні спазми трансформуються в тонічні напади за відсутності латентного періоду і плавно переходять в СЛГ.
2. інфантильні спазми зникають; психомоторне розвиток дитини дещо покращується; картина ЕЕГ поступово нормалізується. Потім настає латентний період, який варіює за тривалістю у різних хворих; з'являються напади раптових падінь, атипові абсанси і наростає дифузна повільна пік-хвильова активність на ЗЗГ.
Для СЛГ характерна тріада нападів: пароксизми падінь (атоніческі- і міо-клонічні-астатические); тонічні напади і атипові абсанси. Найбільш типові напади раптових падінь, обумовлені тонічними, миоклоническими або атоническими (негативний міоклонус) пароксизмами. Свідомість може бути збережене або вимикається короткочасно. Після падіння не спостерігається судом, і дитина відразу ж встає. Часті напади падінь призводять до важкої травматизації та інвалідизації хворих.
Тонічні напади бувають аксіальним, проксимальними або тотальними; симетричними або чітко латералізованние. Напади включають в себе раптове згинання шиї і тулуба, підйом рук в стані полуфлексіі або розгинання, розгинання ніг, скорочення лицьової мускулатури, обертальні рухи очних яблук, апное, гіперемія обличчя. Вони можуть виникати як в денний час, так і, особливо часто, вночі.
Атипові абсанси також характерні для СЛГ. Прояви їх різноманітні. Порушення свідомості буває неповним. Може зберігатися деяка ступінь рухової і мовної активності. Спостерігається гипомимия, слинотеча; миоклонии століття, рота; атонические феномени (голова падає на груди, рот відкритий). Атипові абсанси зазвичай супроводжуються зниженням м'язового тонусу, що викликає як би "обмяканіе" тіла, починаючи з м'язів обличчя і шиї.
У неврологічному статусі відзначаються прояви пірамідної недостатності, координаторні порушення. Характерно зниження інтелекту, що не досягає, однак, тяжкого ступеня. Інтелектуальний дефіцит констатується з раннього віку, передуючи захворюванню (симптоматичні форми) або розвивається відразу після появи нападів (Криптогенні форми).
При ЕЕГ дослідженні у великому відсотку випадків виявляється нерегулярна дифузна, часто з амплітудною асиметрією, повільна пік-хвильова активність з частотою 1,5-2,5 Гц в період неспання і швидкі ритмічні розряди з частотою близько 10 Гц-під час сну.
При нейровізуалізації можуть мати місце різні структурні порушення в корі головного мозку, включаючи вади розвитку: гіпоплазія мозолистого тіла, гемімегаленцефалія, кортикальні дисплазії та ін.
У лікуванні СЛГ слід уникати препаратів, що пригнічують когнітивні функції (барбітурати). Найбільш часто при СЛГ застосовуються вальпроати, ламотриджин, карбамазепін, бензодіазепіни. Лікування починається з похідних вальпроєвої кислоти, поступово збільшуючи їх до максимально переносимої дози (70-100 мг / кг / сут. І вище). Карбамазепін ефективний при тонічних нападах - 15-30 мг / кг / сут, але може учащати абсанси і міоклонічні пароксизми. Ряд хворих реагує на збільшення дози карбамазепіну парадоксальним почастішанням нападів. Бензодіазепіни надають ефект при всіх типах нападів, однак цей ефект тимчасовий. У групі бензодіазепінів застосовуються клоназепам, клобазам (фрізіум) і нитразепам (радедорм). При атипових абсансах може бути ефективний суксилеп (але не як монотерапія). Нами показана висока ефективність комбінації вальпроатов з Ламікталом (1-5 мг / кг / сут. І вище) (табл. 5). Дана терапія найбільш оптимальна як щодо ефективності, так і переносимості (не пригнічує когнітивні функції). У США широко використовується комбінація вальпроатов з фелбаматом (Талокса).
Прогноз при СЛГ важкий. Стійкий контроль над нападами досягається лише у 5-15% хворих. Прогностично сприятливо переважання миоклонических нападів і відсутність грубих структурних змін в мозку; негативні фактори -домінірованіе тонічних нападів і грубий інтелектуальний дефіцит.
Симптоматична парціальна епілепсія
При симптоматичних парціальних формах епілепсії виявляються структурні зміни в корі головного мозку. Причини, що детермінують їх розвиток, різноманітні і можуть бути представлені двома основними групами: перинатальними і постнатальної факторами. Перинатальна енцефалопатія в анамнезі констатується у 35% пацієнтів; серед постнатальних факторів слід зазначити нейроінфекції, черепно-мозкові травми, пухлини кори головного мозку.
Дебют нападів при симптоматичної парціальної епілепсії варіює в широкому віковому діапазоні, з максимумом в предшкольном віці. Для цих випадків характерна наявність змін в неврологічному статусі, нерідко в поєднанні зі зниженням інтелекту; поява регіональних патернів на ЕЕГ, резистентність нападів до АЕП. Виділяють симптоматичні парціальні форми епілепсії: скроневу, лобову, тім'яну і потиличну. Дві перших - найбільш часті і складають до 80% всіх випадків.
Симптоматична скронева епілепсія
Клінічні прояви ВЕ вкрай різноманітні. У ряді випадків атипові фебрильні судоми передують розвитку захворювання. ВЕ проявляється простими, складними парціальними, вторинно генералізованими нападами або їх поєднанням. Особливо характерна наявність складних парціальних нападів, що протікають з розладом свідомості, в поєднанні з збереженій, але автоматизованої руховою активністю. Автоматизми при складних парціальних нападах можуть бути односторонніми, виникаючи на гомолатеральной вогнищу стороні, і часто поєднуються з дистонической установкою кисті на контрлатеральной стороні. ВЕ поділяють на амігдала-гиппокампального (палеокортікальную) і латеральну (неокортикальної) епілепсію.
Амігдала-гиппокампального скронева епілепсія характеризується виникненням нападів з ізольованим розладом свідомості. Спостерігається застигання хворих з маскоподібним особою, широко розкритими "здивованими" очима і заставленим в одну точку поглядом. При цьому можуть констатувати різні вегетативні феномени: збліднення особи, розширення зіниць, пітливість, тахікардія. Виділяють 3 типи СПП з ізольованим розладом свідомості:
вимикання свідомості з завмиранням і раптовим перериванням рухової і психічної активності;
вимикання свідомості без переривання рухової активності;
вимикання свідомості з повільним падінням ( "обмяканіем") без судом ( "скроневі синкопи").
Характерні також вегетативно-вісцеральні пароксизми. Напади проявляються відчуттям абдомінального дискомфорту, болю в області пупка або епігастріума, бурчання в животі, позивів на дефекацію, відходження газів (епігастральні напади). Можлива поява "висхідного епілептичного відчуття", що описується хворими як біль, печія, нудота, що виходить з живота і піднімається до горла, з почуттям стиснення, здавлення шиї, грудки в горлі, нерідко з наступним виключенням свідомості і судомами. При залученні в процес мигдалеподібного комплексу виникають напади страху, паніки або люті; роздратування гачка викликає поява нюхових галюцинацій. Можливі напади з порушенням психічних функцій (сновідних стану, вже баченого або ніколи не баченого і ін.).
Латеральна ВЕ проявляється нападами з порушенням слуху, зору і мови. Характерно поява яскравих кольорових структурних (на відміну від потиличної епілепсії) зорових галюцинацій, а також складних слухових галюцинацій. Близько 1/3 жінок, які страждають ВЕ, відзначають почастішання нападів в періменструальном періоді.
При неврологічному обстеженні дітей, які страждають ВЕ, нерідко виявляються мікроочаговие симптоми, контрлатерально вогнища: недостатність функції 7 і 12 черепних нервів за центральним типом, пожвавлення сухожильних рефлексів, поява патологічних рефлексів, легкі координаторні розлади та ін. З віком у більшості пацієнтів виявляються стійкі порушення психіки , які проявляються переважно інтелектуально-мнестическими або емоційно-особистісними розладами; характерна поява виражених розладів пам'яті. Збереження інтелекту залежить, головним чином, від характеру структурних змін в мозку.
При ЕЕГ дослідженні спостерігається пік-хвильова або частіше стійка регіональна повільнохвильовий (тета) активність в скроневих відведеннях, зазвичай з поширенням вперед. У 70% хворих виявляється виражене уповільнення основний активності фонового запису. У більшості пацієнтів з плином часу епілептична активність відзначається бітемпорально. Для виявлення вогнища, локалізованого в медио-базальних відділах, краще застосування інвазивних сфеноідальние електродів.
При нейровізуалізації виявляються різні макроструктурні порушення в головному мозку. Частою знахідкою при МРТ дослідженні є медіальний скроневий (інцізуральний) склероз. Нерідко також відзначається локальне розширення борозен, зменшення в обсязі залученої скроневої частки, парціальна вентрікуломегалія.
лікуванняВЕ - складне завдання; багато пацієнтів резистентні до терапії. Базові препарати - похідні карбамазепіну. Середня добова доза становить 20 мг / кг. При неефективності -збільшення дози до 30 мг / кг / сут. і вище до появи позитивного ефекту або перших ознак інтоксикації. При відсутності ефекту слід відмовитися від застосування карбамазепіну, призначивши замість нього фенітоїн при складних парціальних нападах або вальпроати при вдруге генералізованих пароксизмах. Дозування дифенина при лікуванні ВЕ становить 8-15 мг / кг на добу, вальпроатов -50-100 мг / кг / сут. При відсутності ефекту від монотерапії можливе застосування политерапии: карбамазепін + вальпроати, карбамазепін + ламіктал, карбамазепін + фенобарбітал, фенобарбітал + дифенин (остання комбінація менш краща і викликає значне зниження уваги і пам'яті, особливо у дітей). На додаток до базової терапії АЕП у жінок можуть застосовуватися статеві гормони, особливо ефективні при менструальної епілепсії. Застосовується оксипрогестерона капронат 12,5% розчин 1-2 мл в / м одноразово на 20-22-й день менструального циклу.
Прогноз залежить від характеру структурного ураження мозку. З віком у більшості пацієнтів наростають стійкі порушення психіки, значно ускладнюють соціальну адаптацію. В цілому, близько 30% хворих, які страждають ВЕ, резистентні до традиційної антиепілептичної терапії і є кандидатами на нейрохірургічне втручання.
Симптоматична лобова епілепсія
Клінічна симптоматика лобової епілепсії (ЛЕ) різноманітна. Захворювання проявляється простими і складними парціальними нападами, а також, що особливо характерно, вдруге генералізованими нападами. Виділяють наступні форми ЛЕ: моторна, оперкулярной, дорсолатеральна, орбітофронтальная, передня фронтополярная, цінгулярной, яка виходить із додаткової моторної зони.
Моторні пароксизми виникають при подразненні передньої центральної звивини. Характерні джексоновские напади, що розвиваються контрлатерально вогнища. Судоми мають переважно клонический характер і можуть поширюватися по типу висхідного (нога-рука-обличчя) або низхідного (особа-рука-нога) маршу; в деяких випадках із вторинною генералізацією. При вогнищі в парацентральних часточках судоми можуть спостерігатися в ипсилатеральной кінцівки або білатерально. Постпріступном слабкість в кінцівках (параліч Тодда) - частий феномен ЛЕ.
Оперкулярной напади виникають при подразненні оперкулярной зони нижньої лобової звивини на стику з скроневої часток. Виявляються пароксизмами жувальних, смоктальних, ковтальних рухів, прицмокуванням, облизування, покахикуванням; характерна гіперсалівація. Можливо іпсилатеральний посмикування м'язів обличчя, порушення мови або мимовільна вокализация.
Дорсолатеральних напади виникають при подразненні верхньої і нижньої лобової звивини. Виявляються адверсівних нападами з насильницьким поворотом голови і очей, зазвичай контрлатерально вогнища роздратування. При залученні задніх відділів нижньої лобової звивини (центр Брока) констатуються пароксизми моторної афазії.
Орбітофронтальної напади виникають при подразненні орбітальної кори нижньої лобової звивини і проявляються різноманітними вегетативно-вісцеральними феноменами. Характерні епігастральні, кардіоваскулярні (болі в області серця, зміна серцевого ритму, артеріального тиску), респіраторні (інспіратор-ва задишка, відчуття задухи, стиснення в області шиї, "кома" в горлі) напади. Нерідко з'являються фарингіт-оральні автоматизми з гіперсалівацією. Звертає на себе увагу велика кількість вегетативних феноменів в структурі нападів: гіпергідроз, блідість шкіри, нерідко з гіперемією особи, порушення терморегуляції та ін. Можлива поява типових складних парціальних (психомоторних) пароксизмів з автоматизмами жестів.
Передні (фронтополярние напади виникають при подразненні полюса лобових часток. Характеризуються простими парціальними нападами з порушенням психічних функцій. Виявляються відчуттям раптового "провалу думок", "порожнечі в голові", розгубленості або, навпаки, насильницьким спогадом; болісним, тяжким відчуттям необхідності щось згадати. Можливий насильницький "наплив думок", "вихор ідей" - відчуття раптової появи у свідомості думок, не пов'язаних за змістом з поточної розумовою діяльністю. Пацієнт ні і меет можливості позбутися від цих думок до закінчення нападу.
Цінгулярной напади виходять з передньої частини поясної звивини медіальних відділів лобових часток. Виявляються переважно складними, рідше простими парціальними нападами з порушенням поведінки і емоційної сфери. Характерні складні парціальні напади з автоматизмами жестів, почервонінням особи, виразом переляку, ипсилатерально моргательнимі рухами, а іноді клонічними судомами контрлатеральний кінцівок. Можлива поява пароксизмальних дисфорических епізодів зі злостивістю, агресивністю, психомоторним збудженням.
Напади, які виходять із додаткової моторної зони, вперше були описані Пенфілдом, але систематизовані лише останнім часом. Це досить частий вид нападів, особливо якщо врахувати, що пароксизми, що виникають в інших відділах лобової частки, нерідко іррадіюють в додаткову моторну зону. Характерно наявність частих, зазвичай нічних, простих парціальних нападів з альтернирующая геміконвульсіі, архаїчними рухами; нападів з припиненням мовлення, нечіткими, погано локалізованими чутливими відчуттями в тулубі та кінцівках. Парціальні моторні напади проявляються зазвичай тонічними судомами, що виникають то з одного, то з іншого боку, чи білатерально (при цьому виглядають як генералізовані). Характерно тонічне напруга з підйомом контрлатеральной руки, адверсія голови і очей (хворий як би дивиться на свою підняту руку). Описано виникнення "гальмівних" нападів з пароксизмальним гемипарезом. Напади архаїчних рухів виникають зазвичай у нічний час з високою частотою (до 3-10 раз за ніч, нерідко щоночі). Характеризуються раптовим пробудженням пацієнтів, криком, гримасою жаху, рухової бурею: розмахуванням руками і ногами, боксування, педалюванням (що нагадує їзду на велосипеді), тазовими рухами (як при коїтус) та ін. Ступінь порушення свідомості флюктуирует, але в більшості випадків свідомість збережена. Дані напади слід диференціювати від істеричних і пароксизмальних нічних страхів у дітей.
ЕЕГ дослідження при ЛЕ може показувати такі результати: норма, пік-хвильова активність або уповільнення (періодичне ритмічне або продовжене) регіонально в лобових, лобно-центральних або лобно-скроневих відведеннях; біфронтальние незалежні пік-хвильові вогнища; вторинна билатеральная синхронізація; регіональна фронтальна низкоамплитудная швидка (бета) активність. Вогнища, локалізовані в орбитофронтальной, оперкулярной і додаткової моторної зоні можуть не давати змін при накладенні поверхневих електродів і вимагають застосування глибинних електродів або кортікографіі. При ураженні додаткової моторної зони ЗЕГ патерни нерідко ипсилатерально нападів або білатеральні, або має місце феномен вторинної білатеральної синхронізації Джаспера.
лікування ЛЕ здійснюється за загальними принципами терапії локалізаціонно-обумовлених форм епілепсії. Карбамазепін і дифенин є препаратами вибору; вальпроати, ламотриджин і барбітурати - резервними. Вальпроати особливо ефективні в разі вторинно генералізованих судомних нападів. При неефективності монотерапії застосовується политерапия - комбінація перерахованих вище препаратів. Повна резистентність нападів до проведеної терапії АЗП є приводом для розгляду питання про оперативне втручання.
Прогноз ЛЕ залежить від характеру структурного ураження мозку. Часті напади, резистентні до терапії, значно погіршують соціальну адаптацію хворих. Напади, які виходять із додаткової моторної зони, зазвичай резистентні до традиційних АЕП і вимагають хірургічного лікування.
Загальні принципи лікування епілепсії
В даний час вироблені загальноприйняті міжнародні стандарти щодо лікування епілепсії, яких необхідно дотримуватися для підвищення ефективності лікування і поліпшення якості життя пацієнтів.
Лікування епілепсії може бути почато тільки після встановлення точного діагнозу. Терміни "предепілепсія" і "профілактичне лікування епілепсії" є абсурдними. На думку більшості неврологів, лікування епілепсії слід починати після повторного нападу. Одиничний пароксизм може бути "випадковим", обумовленим лихоманкою, перегрівом, інтоксикацією, метаболічними розладами і не ставитися до епілепсії. В цьому випадку негайне призначення АЕП не може бути виправданим, так як ці препарати є потенційно високотоксичними і не застосовуються з метою "профілактики". Таким чином, АЕП можуть застосовуватися тільки в разі повторних непровоціруемих епілептичних нападів (тобто при епілепсії за визначенням).
Діагноз епілепсії встановлено, і вирішено призначити АЕП. З 1980-х років в клінічній епілептології міцно утвердився принцип монотерапії: купірування епілептичних нападів повинно здійснюватися переважно одним препаратом. В даний час повністю доведена неспроможність старої концепції про призначення великої кількості АЕП одночасно в малих дозах. Політерапія виправдана тільки в разі резистентних форм епілепсії і не більше 3 АЕП одночасно.
Підбір АЕП не повинен бути емпіричним. АЕП призначаються строго відповідно до форми епілепсії і характером нападів. Успіх лікування епілепсії багато в чому визначається точністю сіндромологіческой діагностики (табл. 1).
АЕП призначаються, починаючи з малої дози, з поступовим збільшенням до досягнення терапевтичної ефективності або появи перших ознак побічних ефектів. При цьому визначальним є клінічна ефективність і переносимість препарату (табл. 2).
У разі неефективності одного препарату він повинен бути поступово замінений іншим АЕП, ефективним при даній формі епілепсії. При неефективності одного АЕП не можна відразу додавати до нього другий препарат, тобто переходити на політерапію, не використовуючи всіх резервів монотерапії.
Показання до визначення змісту АЕП в крові. Визначення рівня АЕП в крові може бути корисно в клінічній практиці в наступних випадках:
Поява ознак інтоксикації;
Відсутність ефективності при застосуванні адекватних доз;
Наявність у пацієнта станів, що викликають порушення фармакокінетики АЕП (захворювання печінки і нирок, вагітність, ранній вік і ін.);
Рекомендується в усіх випадках застосування фенітоїну (нелінійна фармакокінетика);
Застосування пацієнтом декількох АЕП або інших препаратів, що змінюють фармакокінетику АЕП;
Проведення медичної експертизи.
Слід завжди пам'ятати, що між клінічною ефективністю препарату, його переносимість та концентрацією в крові чіткої прямої залежності не існує. У зв'язку з цим клінічний критерій ефективності і переносимості повинен завжди домінувати над лабораторними показниками.
Застосування в лікуванні ІЕ інших препаратів, крім антиепілептичних, - предмет дискусії. У деяких з обстежених нами хворих в комплексній терапії були використані судинні (циннарізін, кавінтон, сермион, танакан); ноотропні (ноотропіл, пантогам, когітум, оксибрал); метаболічні (есенціале-форте, фолієва кислота) препарати. Вплив їх на частоту нападів і загальне самопочуття пацієнтів не аналізувалося. На думку абсолютної більшості зарубіжних дослідників, препарати даних груп не мають жодного впливу на перебіг епілепсії та не застосовуються при даному захворюванні. Разом з тим, в окремих випадках при поєднанні епілепсії з головними болями, порушеннями пам'яті і концентрації уваги, токсичну дію АЕП, застосування судинних, ноотропних і метаболічних препаратів може бути виправдане. Однак немає виправдання "патологічного" завзятості деяких лікарів, що призначають пацієнтам по 5-8 різних препаратів типу сечогінних, розсмоктуючих, вітамінних і т.д. без будь-якого здорового глузду. Призначення будь-якого препарату повинно бути строго аргументовано.
Принципи скасування лікування
АЕП можуть бути відмінені через 2,5-4 року повної відсутності нападів. Клінічний критерій (відсутність нападів) є основним критерієм скасування терапії. При більшості идиопатических форм епілепсії скасування препаратів може здійснюватися через 2,5 (роландична епілепсія) - 3 роки ремісії. При важких резистентних формах (синдром Леннокса-Гасто, симптоматична парціальна епілепсія), а також при юнацької міоклонічні епілепсії, даний період збільшується до 3-4 років. При тривалості повної терапевтичної ремісії протягом 4 років, лікування повинно бути скасовано у всіх випадках. Наявність патологічних змін на ЕЕГ або пубертатний період пацієнтів не є чинниками, що затримують скасування АЕП при відсутності нападів більше 4 років.
К.В. Воронкова, д. М. Н., Професор, ГБОУ ВПО «Російський національний медичний університет ім. Н.І. Пирогова »МОЗ РФ, м.Москва Ключові слова: Епілепсія, діагностика епілепсії, типи нападів, форми епілепсії у дітей, фебрильні судоми.
Key words: Epilepsy, diagnosis of epilepsy, seizure types, forms of epilepsy in children, febrile convulsions.
Вступ
Епілепсія - група часто зустрічаються захворювань головного мозку, що виявляються нападами. У світі понад 60 млн осіб страждають на епілепсію, в цілому в популяції - приблизно 0,5-1%. У Росії - близько 2 млн дітей і дорослих, а в США - 2,7 млн хворих. За даними деяких досліджень, епілепсія частіше зустрічається у чоловіків, ніж у жінок.
Епілепсія - одне з найпоширеніших захворювань в дитячому віці, яке зустрічається частіше, ніж, наприклад, бронхіальна астма або туберкульоз. В 2/3 всіх випадків епілепсія починається до 20-річного віку. В цілому епілепсії у дітей розглядаються як доброякісні стану, які можуть проходити самостійно або на тлі терапії в пубертатному віці, проте це не так. Багато епілепсії дитячого віку можуть важко піддаватися лікуванню і супроводжуватися серйозними порушеннями у вищій психічній сфері. Багато епілепсії у дітей вчасно не розпізнаються і не лікуються. Може спостерігатися і гіпердіагностика, коли в епілепсію екстраполюються такі стани, як енурез, порушення поведінки та ін. Все це має негативні наслідки для дитини.
Необхідно також відзначити, що дитяча епілептологія надзвичайно багатолика, зустрічаються форми, що не відзначаються у дорослих. Крім того, у дітей на прояви епілепсії впливають безліч специфічних факторів. Саме у дітей виділено цілий ряд форм епілепсії з дебютом в певному віці (возрастзавісімих форми епілепсії) - від періоду новонародженості до підліткового віку. Вік дебюту вказано в назві даних форм епілепсії. Наприклад, доброякісні судоми новонароджених, дитяча абсанс-епілепсія, доброякісна міоклонічна епілепсія дитинства та ін. Для цих форм захворювання вік початку нападів - один з важливих ознак, що дозволяють поставити правильний діагноз. Багато проявів епілепсії можуть змінюватися з віком і бути пов'язаними з порушенням дозрівання головного мозку. Так, наприклад, інфантильні спазми (синдром Веста) - напади у вигляді «кивків», «складань» ( «салаамови напади»), як правило, починаються в перші 6 місяців життя і спостерігаються тільки в перші 1-1,5 року життя. У дітей старше 1-2 років ці напади проходять або трансформуються в інші: напади падінь, напади «завмирань», тонічні і тоніко-клонічні судомні напади та ін. У таких хворих часто відзначається порушення інтелекту. З іншого боку, типові абсанси, що починаються в дитячому або юнацькому віці (в рамках дитячої та юнацької абсанс-епілепсій), - ознака більш «доброякісних» форм епілепсії, як правило, не супроводжуються зниженням інтелекту і іншими порушеннями з боку нервової системи. Вік дебюту і протягом даних форм епілепсії, включаючи можливу еволюцію, - процес генетично детермінований.
Дана стаття актуалізує проблему дитячих епілепсій в широкій медичній практиці, Оскільки з проявами епілепсії можуть зіткнутися не тільки неврологи і психіатри, а й лікарі різних спеціальностей, і особливо - педіатри. Безсумнівно, завданням педіатра є лише розпізнати серед патологічних симптомів прояв епілепсії і відправити дитину на консультацію до фахівця.
Наведемо два клінічних прикладу про складнощі диференціальної діагностики дитячої епілепсії. Обидва приклади свідчать також про те, що можливо запізнювання постановки діагнозу, який з об'єктивних причин може бути поставлений з плином часу і появою додаткових «доказів». Те ж стосується не тільки постановки діагнозу епілепсії як такого, але і встановлення форми. Іноді потрібно багато років, щоб форма захворювання стала очевидна в зв'язку з можливою трансформацією і еволюцією епілепсії, що також частіше зустрічається у дітей, ніж у дорослих.
Дівчинка, 12 років . З 8 років турбували рідкісні запаморочення. Після дебюту запаморочень через кілька місяців стали з'являтися пароксизмальні стану, що починаються з запаморочення і страху з подальшою адверсія голови і тулуба вправо і падінням, дівчинка при цьому перебувала у свідомості. Тривалість таких станів становила 1-5 хвилин, а частота - 1 раз на тиждень або місяць. Спостерігалися у невролога за місцем проживання кілька років з діагнозом «вегетативна дисфункція», призначалася ноотропні і судинна терапія, але без позитивного ефекту. У 12 років відзначено почастішання пароксизмів (до декількох разів на тиждень), особливо при недосипанні і хвилюванні, в задушливому приміщенні. Також з'явилися генералізовані тоніко-клонічні судомні напади з версівним компонентом з наступною блювотою і тривалим постпріступном сном. При нейровізуалізації виявлена дифузна зона зміненого сигналу в сірій речовині правої тім'яної частки. Було проведено відео-ЕЕГ-моніторинг на тлі депривації сну, в задушливому приміщенні і на тлі стресу, під час якого розвинувся статус адверсівних фокальних епілептичних нападів, що супроводжувалися на ЕЕГ іктальнимі паттернами. Дівчинка була госпіталізована в стаціонар, де була діагностована «симптоматична лобновісочная епілепсія, статусне протягом» і підібрана протисудомна терапія (вальпроєва кислота), після чого напади стали рідшими, статусу епілептичних нападів більше не повторювалося.
Дівчинка, 10 років . З раннього дитинства спостерігалася у невролога з приводу синдрому гіперактивності з дефіцитом уваги. Дебют захворювання відзначений в 9 років, уві сні вночі з'явилося тонічне напруження лівої половини м'язів обличчя з приєднанням клонических посмикувань м'язів кінцівок. Аналогічний напад повторився через 2 місяці, але він був більш тривалим (до 10 хвилин). Більшість нападів приурочені до сну. Дівчинці було проведено нічний відео-ЕЕГ-моніторинг, під час якого були зафіксовані т. Н. доброякісні епілептиформні розряди дитинства в лівій і правій центрально-скроневої і заднелобних областях. Була діагностована «доброякісна епілепсія дитячого віку з центральнотемпоральнимі спайками, роландична». Після призначення протисудомної терапії (вальпроєва кислота) напади більше не повторювалися, покращилася концентрація уваги, здатність до навчання, знизилася емоційна лабільність, дівчинка стала краще встигати в школі.
Характеристика групи епілепсій
визначення епілепсії. Епілепсія - це хронічне захворювання головного мозку, що характеризується повторними непровоціруемимі нападами порушень рухових, вегетативних, сенсорних і психічних функцій, що виникають внаслідок надмірних нейронних розрядів. Це визначення дане головним регламентує дію лікаря-епілептології органом - Міжнародною лігою по боротьбі з епілепсією (International League Against Epilepsy - ILAE), створеної більше 100 років тому.
З визначення випливає кілька висновків:
- Дуже важливий критерій повторності клінічного події, тільки на який і може спиратися лікар, ставлячи діагноз «епілепсія». Однак є ситуації, коли діагноз ставиться після вперше виник епізоду.
- Епілепсія - це спонтанні (непровоціруемие) напади. Напади, що виникають тільки при дії провокуючих чинників, є ситуаційно обумовленими: лихоманка у маленьких дітей (фебрильні судоми), метаболічні порушення (наприклад, при цукровому діабеті), Вживання алкоголю або наркотиків тощо. До цієї ж категорії відносять напади, що виникають на тлі захворювань головного мозку (нейроінфекцій, судинних уражень, черепно-мозкової травми). Виняток становлять рефлекторні форми епілепсії, які характеризуються специфічними способами провокації нападів. До них відносяться: фотосенсітівная епілепсія (при якій приступи викликаються ритмічним миготінням світла), епілепсія читання, епілепсія їжі, провокація нападів тактильним роздратуванням, різними іграми, певними розумовими процесами і ін.
- Крім генералізованих тоніко-клонічних нападів (традиційно асоціюються з поняттям «епілепсія»), описано безліч різних типів нападів. Наприклад, напади у вигляді порушення зорового сприйняття (зорових ілюзій), короткочасного порушення пам'яті, відчуття страху, насильницьких думок, напади у вигляді болю в животі, блювання, задухи і інші, які не супроводжуються судомами. Якщо у хворого, особливо у дитини, виникають такі напади, лікарі нерідко протягом багатьох років не можуть розпізнати їх епілептичну природу. Нерідко діагноз встановлюється тільки після приєднання судомних нападів як результату прогресування хвороби, що видно з наведених клінічних прикладів. При деяких формах епілепсії у одного хворого можуть відзначатися різні типи нападів.
- В основі всіх клінічних проявів епілепсії завжди лежить патологічний, надмірно сильний розряд в нервових клітинах кори великих півкуль головного мозку, який може бути зареєстрований за допомогою електрофізіологічних методів діагностики - електроенцефалографії, або ЕЕГ.
діагностика епілепсії
Діагноз «епілепсія» складається з трьох компонентів (так званий клініко-електро-анатомічний підхід в діагностиці).
- Клінічні прояви нападів (нижче більш детально розберемо напади і часто зустрічаються в практиці дитячого невролога форми епілепсії) і анамнез.
- Зміни біоелектричної активності мозку (електроенцефалографія - ЕЕГ, відео-ЕЕГ-моніторинг). ЕЕГ-дослідження має бути проведено всім хворим з підозрою на епілепсію. На ЕЕГ хворого на епілепсію можливе виявлення епілептиформних змін (у вигляді комплексів «спайк-хвиля» або «пік-хвиля», окремих спайки, поліспайк, гострих хвиль, комплексів «гостра хвиля - повільна хвиля» та ін.), Які відповідають локалізації та поширенню патологічного розряду в корі.
Причому найбільш інформативним і сучасним методом є тривала запис ЕЕГ в різних функціональних станах (неспання, сон) з відеореєстрації відбувається з пацієнтом - відео-ЕЕГ-моніторинг. Відео-ЕЕГ-моніторинг дозволяє не тільки здійснювати більш точну діагностику епілепсії, але також і відстежувати результати лікування.
В даному розділі необхідно розглянути термін «знижена судомна готовність», який часто описують на енцефалограмі, що призводить до гіпердіагностики і призначенням протисудомної терапії у випадках, коли епілепсії у дитини немає. В даний час цей термін вважається некоректним. Необхідно більш точний опис виявлених порушень. У більшості випадків лікування призначається тільки тоді, коли у хворого виникають напади, що супроводжуються епілептиформні активністю на ЕЕГ. Як правило, якщо зміни на ЕЕГ виявляються, а клінічні прояви нападів відсутні, лікування не повинно призначатися. При проведенні ЕЕГ з приводу будь-яких скарг (таких як головний біль і ін.), Але без клінічних ознак епілепсії епілептиформна активність виявляється в 2-3% випадків.
Однак існують стану з так званої групи епілептичних енцефалопатій, коли епілептиформна активність сама по собі робить негативний вплив на пізнавальні функції дітей і може взагалі не проявлятися епілептичними нападами. Особливе значення відіграє дослідження під час сну, коли епілептиформні розряди заміщають фізіологічну біоелектричну активність. Від розрядів страждають нейрональні мережі, відповідальні за утримання інформації, побудови програми дій. Діти можуть втрачати набуті навички, втрачати мова, ставати гіперактивними, аутистичності, а заняття з такими пацієнтами стають малоефективними. Такі форми захворювання були відкриті в останні роки. До них відносяться: синдром Ландау - Клеффнера (придбана епілептична афазія), ESES (електричний статус повільно хвильового сну), аутистический епілептиформний регрес і ін. Такі стани ще називають когнітивними дезінтеграції. У цих виняткових випадках зміни картини ЕЕГ, навіть за відсутності нападів, служать показанням для розгляду призначення антиепілептичних препаратів. - Зміни структури головного мозку, які виявляються методами нейровізуалізації - комп'ютерна томографія (КТ), магнітно-резонансна (МР) візуалізація, в тому числі Високороздільна МРТ в режимі епілептологіческіх сканування. За свідченнями застосовуються і інші методики.
- В окремих випадках за показаннями проводиться генетичне дослідження і аналізи на біохімічні маркери захворювань з групи обмінних порушень (аміноацідопатіі, мітохондріальні, пероксисомні захворювання та ін.).
Розглянемо прояви епілепсії, які можуть відзначатися у дітей, - напади. Слово «напад» іноді використовується для опису інших короткочасних порушень, не пов'язаних з епілепсією: непритомність, психогенні напади, нічні страхи, спалахи гніву та ін. Тому бажано використовувати термін «епілептичний напад», якщо мова йде про епілепсію. З іншого боку, в медичній лексиці існує і багато інших термінів, також використовуються для опису епілептичних нападів - іктальние епізоди, припадки, судоми, конвульсії, пароксизми, епізоди «відключення» (свідомості), атаки та ін.
Епілептичний напад - це короткочасний епізод патологічних надмірних розрядів нервових клітин кори головного мозку, що викликає стан, помітне або непомітне для людини, у якого виникає напад, або для оточуючих людей.
Клінічні прояви нападів дуже різноманітні у різних хворих. Епілептичні напади часто мають раптовий початок і зазвичай припиняються спонтанно, тобто самостійно. Вони, як правило, короткі, тривалістю від декількох секунд до декількох хвилин, і часто супроводжуються періодом сонливості чи сплутаності свідомості (постпріступном період). Існують загальноприйняті класифікації епілептичних нападів - Кіото, 1981 року народження, і розроблені вже в XXI столітті.
Перш за все виділимо такі поняття, як фокальний або вогнищевий напад і генералізований. Фокальні епілептичні напади - це напади, що починаються з однієї півкулі. Прості (без втрати свідомості) і складні (з втратою свідомості) фокальні напади в даний час не виділяються. Проте пропонується оцінювати порушення свідомості в момент нападу. Фокальні напади можуть трансформуватися у вторинно-генералізовані. У такому випадку говорять про ауру (аура - це, власне, початок нападу). Аура має дуже важливе значення, так як вказує на локальний характер нападу і дозволяє припустити локалізацію епілептичного вогнища в корі головного мозку. Виявлення фокальних нападів вимагає з'ясування їх етіології (походження). Генералізований епілептичний напад - напад, що починається одночасно з двох півкуль мозку. Генералізовані напади діляться на судомні і бессудорожние і значно варіюють по клінічно проявів і ступеня тяжкості.
Клінічні прояви фокального нападу залежать від області кори, в якій виникає розряд. Прикладами можуть служити локальні судоми (наприклад, в особі, в руці), вегетативні порушення (почервоніння або блідість обличчя, біль, задишка, посилене потовиділення, запаморочення, нудота і навіть багатогодинна ізольована блювота), а також порушення чутливості (незвичайні зорові, слухові, смакові, нюхові, тактильні відчуття, ілюзії чи галюцинації) або психічні розлади (незвичайні думки, страхи, агресія, зміна реальності того, що відбувається і ін.).
Хворий під час фокального нападу з втратою свідомості може смикати одяг, вертіти в руках предмети, безцільно бродити ( «автоматичне» поведінку, або «автоматизми»). Цмокання губами або жувальні рухи, бурмотіння, гримасничанье, роздягання і здійснення безцільних дій можуть спостерігатися ізольовано або в різних комбінаціях. В окремих випадках можлива поява зовнішньої агресії і аутоагресії. Особливо слід підкреслити, що пацієнт виконує всі це мимоволі, в стані зміненої свідомості, не пам'ятає про звершення.
Фокальні напади з вторинною генералізацією (вторинно-генералізовані напади) - це фокальні напади, при яких епілептичний розряд поширюється на кору обох півкуль головного мозку з розвитком генералізованого нападу з появою судом (частіше - тоніко-клонічних, рідше - інших видів судом), втратою свідомості , іноді - з прикушення мови, можливим нетриманням сечі (а іноді і калу). Якщо розряд поширюється дуже швидко, то за зовнішніми проявами вторинно генералізований напад буває важко відрізнити від первічногенералізованного. У цих випадках тільки ЕЕГ (відео-ЕЕГ-моніторинг) допоможе уточнити тип нападу. Іноді так звані постпріступном симптоми випадання можуть сигналізувати лікаря про фокальному характер нападу.
У новій класифікації докладно описані нові типи фокальних нападів.
Наприклад, геластіческіе напади частіше описуються при гамартома гіпоталамуса і проявляються коротким або тривалим (аж до статусу) сміхом в поєднанні з фокальними нападами з руховими симптомами, миоклониями, аксіальним тонічними нападами і флексорного спазмами. Найбільш часто напади дебютують до 5 років, а в разі асоціації з процесами в лобових або скроневих долях головного мозку дебют відзначається після 5 років. ЕЕГ не специфічна (межпріступние і іктальная) - можуть відзначатися регіональні спайки, генералізовані спайк-хвильові розряди.
Генералізовані тоніко-клонічні напади (ГТКП) - найбільш часто зустрічається тип генералізованих нападів. Раніше вони позначалися терміном «припадки grand mal», або «великі судомні напади», які більшості людей і відомі як епілепсія. Перед нападом можливий період у вигляді загального дискомфорту, нездужання, іноді триває годинами. Таким чином, хворий може відчувати, що напад виникне скоро, але не може передбачити точно час його розвитку. Судоми зазвичай припиняються через кілька хвилин. Після того як напад закінчується, починається постпріступном період, іноді може розвинутися сонливість, сплутаність свідомості, головний біль, нудота, блювота. Після припинення судом часто настає глибокий сон, який іноді може неправильно інтерпретуватися як несвідомий стан. При пробудженні хворі не пам'ятають про те, що трапилося; часто відзначається сонливість і поширені ниючі болі в м'язах, пов'язані з надмірною м'язовою активністю під час нападу.
Абсанси раніше називалися нападами petit mal (малі напади). Напади цього типу починаються в дитячому та підлітковому віці. Як правило, вони не зустрічаються в інших вікових групах. Поява абсансов у дорослих вимагає виключення фокальних нападів (так як іноді фокальні напади можуть нагадувати абсанси за своїми зовнішніми проявами). При розвитку нападу хворий раптово завмирає, погляд здається порожнім, відсутнім (простий абсанс); можливо тремтіння повік, ковтання і закидання голови або падіння голови на груди, почервоніння або блідість обличчя, автоматичні руху (складний абсансах). Напади дуже короткі (тривають кілька секунд), і як хворий, так і оточуючі можуть не помічати їх. Дорослі можуть інтерпретувати ці епізоди як погану звичку, особливості характеру, неуважність дитини; також їх називають «задумами» (дитина часто «замислюється») і ін. Іноді абсанси виявляються випадково при обстеженні дитини з приводу зниження шкільної успішності. Це пов'язано з тим, що часті абсанси призводять до порушення концентрації уваги, а через часті «відключень» дитина пропускає навчальний матеріал, не дивлячись на те, що знаходиться в класі. При абсансах на ЕЕГ виявляються дуже характерні зміни (генералізовані пік-хвильові розряди з частотою 3 Гц і більше). Вони можуть провокуватися гипервентиляцией (глибоке ритмічне дихання) під час запису ЕЕГ. Описана картина відповідає типовим Абсанс. Напади цього типу зустрічаються при формах епілепсії з хорошим прогнозом. В цілому інтелект таких дітей не знижений.
Атипові абсанси відрізняються менш раптовим початком і закінченням, свідомість порушено, але не відключено повністю, дитина виглядає загальмованим. Зміни на ЕЕГ під час нападу також відрізняються від таких при типових абсансах (повільна пікволновая активність частотою менше 3 Гц (частіше 2,5-1,5 Гц), дифузна або генералізована, але зазвичай асиметрична, іноді з фокальним початком, групові розряди часом носять нерегулярний характер з явищами флуктуації свідомості). Атипові абсанси характерні для важких форм епілепсій з поганим прогнозом, таких як синдром Леннокса - Гасто, симптоматична лобова епілепсія.
Міоклонічні напади - дуже короткі, раптові, мимовільні скорочення, які можуть залучати все тіло або його частину, наприклад руки або голову. Іноді міоклонічні напади можуть бути причиною падіння пацієнта, проте після падіння напад відразу припиняється. При міоклонічних нападах, які залучають руки, хворі несподівано відкидають предмети (наприклад, хворий відкидає зубну щітку, коли чистить зуби вранці, або столові предмети за сніданком). Часто міоклонічні напади розвиваються в ранкові години. Хворі і родичі не звертаються до лікаря, бачачи подібні «посмикування», пояснюючи їх як прояви «неврозу». І часом тільки приєднання генералізованих тоніко-клонічних нападів з падіннями призводить хворого до лікаря, однак на прийомі такі хворі можуть і не повідомити про попередні нападах «сіпання», і лікар в такому випадку може проявити наполегливість і сам розпитує про додаткові патологічних симптомах.
У новій класифікації виділено новий тип генералізованого епілептичного міоклонусу - масивний білатеральний миоклонус: поодинокі або згруповані в серію аксіальні здригання всього тіла, в основному верхніх кінцівок, що супроводжуються генералізованої полі / спайк-хвильовим розрядом. Спостерігається при юнацької міоклонічні епілепсії, при синдромі Драве.
Міоклонія століття з абсансами - скорочення століття, що провокують в основному закриттям очей і фотостимуляцією при симптоматичних і идиопатических формах епілепсії. Передбачається, що міоклонічні скорочення століття з абсансами можуть бути самостійною, генетично детермінованої формою епілепсії, що дебютує в дитячому віці з тривалим або довічним плином.
Міоклонія століття без абсанса - виникає в перші секунди розряду на ЕЕГ і включає більше трьох скорочень століття, підведення очей вгору, вертикальні їх посмикування, посмикування брів, голови, може спостерігатися девіація голови і очей. Іноді спостерігається миоклония в руках.
Епілептичний негативний міоклонус - зупинка м'язової активності (без попередньої миоклонии), асоційованої з часу зі Спайком на ЕЕГ в поєднанні з фокальними, тонічними, генералізованими тоніко-клонічними або атоническими нападами, абсансами, спазмами. Спостерігається при роландической епілепсії, електричному статус повільного сну, прогресуючих миоклонус-епілепсія, при симптоматичних епілепсія. Може спостерігатися як ускладнення антиепілептичної терапії, і тоді це явище носить назву «агравація епілепсії». Необхідно відзначити, що негативний міоклонус, по суті, є фокальним інгібіторним приступом, а не на генералізований.
Міоклонічно-атонічний напад (раніше - міоклонічно-астатичний) - варіант миоклонии, коли за вздрагиванием слід втрата м'язового тонусу при міоклонічно-астатической епілепсії. Здригання асоційоване з генералізованим спайковую розрядом, атонія - з генералізованою повільної хвилею.
Атонічні і тонічні напади - більш рідкісні типи нападів (менше 1% всіх нападів при епілепсії). Вони зустрічаються при деяких важких формах епілепсії з початком в ранньому дитинстві, таких як синдром Леннокса - Гасто. Атонічні напади проявляються раптової втратою тонусу м'язів, хворий «обм'якає» і падає (повільно «осідає»). Судомні скорочення відсутні. Під час тонічних нападів м'язовий тонус, навпаки, підвищується, м'язи стають дуже напруженими ( «як кам'яні»), що також супроводжується падінням хворого, в результаті якого можливе отримання травми. Тонічні і атонічні напади - часто результат важкого ураження мозку. Пропонується виділяти два варіанти атонических нападів - короткий (з короткою втратою свідомості або без неї з швидким вставанням після атонії) і пролонгований (втрата свідомості і генералізована атонія тривають до декількох хвилин).
Форми епілепсії дитячого віку
Необхідно відзначити, що в дитячому віці можуть зустрічатися практично всі форми епілепсії. Загальноприйнятою і доступною є Міжнародна класифікація епілепсій та епілептичних синдромів (1989 г.), в яку покладено два основних принципи, згідно з якими всі епілепсії та епілептичні синдроми підрозділяються по етіології (ідіопатичні, симптоматичні, Криптогенні) і за характером (парціальні (фокальні) і генералізовані ) нападів. Крім того, враховується вік дебюту, провідний тип нападу і прогноз. У XXI столітті пропонуються нові підходи до класифікації епілепсії - це каталог відомих, вікова класифікація та ін.
Чи не станемо детально зупинятися на фокальних симптоматичних формах епілепсії, які найбільш часто зустрічаються у дорослих хворих на епілепсію, але також часто зустрічаються у дітей. Семіологія нападів при цих формах добре відома і може корелювати з локалізації патологічного процесу і поширенням процесу і епілептиформні активності на інші зони головного мозку.
Судомні напади, що виникають у маленьких дітей (від 6 місяців до 6 років) на тлі високої температури, Називаються фебрильними судомами. Такі напади не розглядаються як епілепсія і відносяться до ситуаційно-обумовленим нападів. Це пов'язано з тим, що напади в даному випадку виникають тільки при певній ситуації (підйом температури) і тільки в певному віці. Спонтанні напади (не пов'язані з температурою тіла) в цих випадках виникати не повинні. Обстеження (що включає огляд невропатолога, оцінку розвитку дитини і ЕЕГ поза нападу) не виявляється патологічних змін. Таким дітям тривала антиепілептичної терапія не призначається, хоча існує велика ймовірність повторення фебрильних судом при наступних захворюваннях дитини, що супроводжуються лихоманкою. Рекомендується уникати підйому температури, застосовувати методи медикаментозного та немедикаментозного зниження температури на тлі інфекційних захворювань; і в деяких випадках препарати для лікування епілепсії призначаються з профілактичною метою на період гострих інфекційних захворювань. З віком (після 5-6 років) фебрильні судоми проходять безслідно. Однак в деяких випадках фебрильні напади є першим проявом епілепсії і надалі супроводжуються появою спонтанних епілептичних нападів, не пов'язаних з лихоманкою. Кордон між фебрильними судомами, що не вимагають лікування, і на епілепсію, що вимагає призначення тривалої антиепілептичної терапії, визначає лікар.
Рання дитяча епілептична енцефалопатія з Супресивна-вибуховими змінами на ЕЕГ - синдром Отахара (СО). При СО в більшості випадків відзначається пренатальне ураження головного мозку. Захворювання починається у віці до 3 місяців, частіше - на 1-му місяці життя дитини. Основний тип нападів - тонічні спазми тривалістю в межах 10 секунд. Вони частіше виникають у вигляді серій (в одній серії може відзначатися по 10-40 спазмів). Загальна кількість спазмів може досягати 300-400 на добу. Також спостерігаються короткі фокальні напади. Відзначаються виражена затримка психічного і моторного розвитку, порушення в неврологічному статусі. Характерні стійкість до терапії і несприятливий прогноз захворювання з високою смертністю в дитячому віці. Можлива трансформація в синдром Веста.
Синдром Веста (СВ) . У новій класифікації відноситься до групи возрастзавісімих епілептичних енцефалопатій дитячого віку. СВ характеризується наступними критеріями: особливим типом епілептичних нападів - інфантильними спазмами, змінами на електроенцефалограмі у вигляді гіпсарітмія, затримкою психомоторного розвитку. З віком більшість випадків СВ трансформуються в фокальні форми епілепсії, рідше - в синдром Леннокса - Гасто. Виявляються зміни: затримка психічного і моторного розвитку.
Синдром Леннокса - Гасто (СЛГ) . Ця форма епілепсії за новою класифікацією відноситься до групи епілептичних енцефалопатій дитячого віку. Захворювання дебютує у віці від 2 до 8 років з піком початку в 3-5 років. Приблизно в 20% випадків СЛГ трансформується з синдрому Веста. Фебрильні судоми передують розвитку синдрому в 10% випадків. При СЛГ відзначаються різноманітні напади: тонічні аксіальні, міатоніческіе падіння, атипові абсанси, епілептичний статус «малих моторних нападів», міоклонічні, генералізовані судомні, фокальні. Епілептичний статус виникає у 75% хворих СЛГ. Часто можуть відзначатися розлади інтелекту.
Доброякісна епілепсія дитячого віку з центрально-скроневими спайками - роландична епілепсія . Починається ця форма епілепсії у віці 3-14 років (в 85% випадків - у віці 4-10 років). Переважають фарингіт-оральні пароксизми: горлові звуки типу «булькання», гіперсалівація (підвищене слиновиділення), анартрия (порушення мови) при збереженому свідомості. При цьому можливо відчуття «поколювання» в області неба, слизової рота; сіпання губи або мови. Нерідко виникають фокальні моторні клонічні напади з локалізацією в лицьової мускулатури, а також в руці, рідше - з залученням ноги на тій же стороні і вторічногенералізованние. Напади рідкісні і нетривалі. Характерно виникнення їх при пробудженні або засипанні. Інтелект не страждає. У деяких дітей відзначається зниження концентрації уваги з гіперактивністю. На ЕЕГ виявляються специфічні знаки - доброякісні епілептиформні розряди дитинства (Дерден) з локалізацією в центрально-скроневих відведеннях. Характерно наростання епілептиформні активності в період повільного сну. Прогноз частіше сприятливий, проте може відбуватися і атипова еволюція в епілептичний статус медленноволнового сну (описаний вище).
Доброякісні потиличні епілепсії дитинства . Існує 2 самостійних синдрому: з раннім (3-6 років) дебютом - варіант Панайотопулоса і пізнім (зазвичай 6-14 років) початком - варіант Гасто. Захворювання проявляється ізольованою зорової аурою, вегетативно-вісцеральними симптомами (може бути нудота, блювота і інші симптоми), фокальними моторними нападами. Характерно виникнення нападів після засипання і перед пробудженням. З'являється головний біль, блювота (ці симптоми також можливі в кінці нападу), адверсія (поворот) очей і голови в бік; геміклоніческіе судоми (судоми однієї половини тіла); можлива вторинна генералізація. Синдром Панайотопулоса проявляється нападами з тривалою втратою свідомості. У дітей старшого віку виникають зорові відчуття - елементарні зорові галюцинації ізольовано або перед початком «великого нападу» або при переході в інші типи нападів. Частота нападів невисока.
Синдром Ландау - Клеффнера (СЛК), або «синдром хронічної епілептичної афазії», за новою класифікацією відноситься до групи епілептичних енцефалопатій дитячого віку. СЛК починається у віці 3-7 років. До моменту дебюту захворювання рухове, психічний і мовний розвиток пацієнтів відповідає віку. Мовні порушення розвиваються поступово, починаючись з порушень розуміння мови, потім приєднується моторна афазія. При СЛК можуть відзначатися епілептичні напади: фокальні моторні напади, атипові абсанси, атонічні, міоклонічні та вторинно генералізовані судомні пароксизми. У 25% хворих СЛК протікає при відсутності епілептичних нападів або наявності одиничних в анамнезі. У неврологічному статусі осередкові симптоми відсутні. На ЕЕГ епілептиформні порушення виявляються в 100% випадків. Морфологія епіактівності відповідає Дерден. Епілептиформна активність наростає уві сні. Прогноз при СЛК щодо епілептичних нападів: практично у 100% пацієнтів напади повністю купіруються і епілептиформна активність блокується до початку пубертатного періоду (під дією АЕП або спонтанно). А прогноз, що стосується мовних, когнітивних і поведінкових порушень, не настільки сприятливий.
Дитяча абсанс-епілепсія (ДАЕ) . Дебют абсансов при дитячій абсанс-епілепсії спостерігається у віковому інтервалі від 3 до 9 років. У дівчаток ця форма епілепсії зустрічається частіше. Для ДАЕ більш характерні складні абсанси, особливо з рухами голови (легке закидання і сіпання), закладом очей вгору тривалістю від 3 до 30 секунд (зазвичай 5-15 секунд) і частотою - десятки і сотні на добу. Перед закінченням нападу у пацієнтів можуть спостерігатися короткі автоматизми (прицмокування, перебирання руками і ін.). Гіпервентиляція (глибоке і часте дихання) - основний провокуючий фактор виникнення абсансов. Генералізовані судомні напади спостерігаються приблизно у 25% хворих ДАЕ.
Юнацька абсанс-епілепсія (ЮАЕ) . Дебют абсансов при юнацької абсанс-епілепсії - частіше в 9-13 років. Абсанси при ЮАЕ зазвичай коротше за тривалістю і рідше по частоті, ніж при ДАЕ. Характерно переважання простих абсансов, тобто без моторного компонента, тривалістю близько 6 секунд. У 65-90% випадків у пацієнтів з ЮАЕ відзначаються генералізовані судомні напади.
Епілепсія з ізольованими генералізованими судорожними нападами . Дебют захворювання спостерігається в широкому віковому діапазоні. Клінічно проявляється єдиним типом нападів - генералізованими судорожними нападами.
Юнацька міоклонічна епілепсія (ЮМЕ, синдром Янца) . Дебют юнацької міоклонічні епілепсії відзначається в віковому інтервалі 11-15 років. Частіше страждають дівчата. Головний симптом - міоклонічні напади, які локалізуються головним чином в плечовому поясі і руках, переважно в розгинальних групах м'язів, рідше захоплюють м'язи ніг. Свідомість під час міоклонічних нападів збережено. Характерно виникнення або збільшення частоти нападів в перші хвилини і години після пробудження пацієнтів. У 90% випадків відзначаються також ГТКП пробудження і в 30% - абсанси. Провокують напади фактори при ЮМЕ - депривація сну, раптове насильницьке пробудження, ритмічне мерехтіння світла й ін.
На закінчення необхідно відзначити, що дитячі епілепсії дуже різноманітні. Необхідно з увагою ставитися як до пароксизмальних симптомам, так і до різних варіантів порушень у вищій психічній сфері. При будь-яких підозрах на епілепсію необхідно направляти дитину на консультацію та обстеження до профільного фахівця.
Список літератури: