Epilepsija i napadi panike. Lečenje epilepsije kod dece. Oblici epilepsije u djetinjstvu
Epilepsija debituje uglavnom u detinjstvu (oko 75% svih slučajeva). Dečju epilepsiju karakteriše veliki broj napadaja otpornih na tretman i polimorfizam napadaja, i što je najvažnije, u detinjstvu se mogu sakriti mnogi opskurni bolni napadi, pupčana kolika, nesvestica, acetonemska povraćanje.
leptičke napade organske prirode, kao što su naznačili Jackson, Specht i Livingston. Ipak, epilepsija ne bi trebala biti dijagnoza isključivanja u slučajevima kada nije pronađeno drugo dijagnostičko objašnjenje. Djeca koja pate od epileptičkih napada brzo razvijaju funkcionalna oštećenja, koja se zatim pretvaraju u trajne promjene karaktera, pamćenja, pažnje, ponašanja i školskog uspjeha.
Dijagnoza i liječenje rezistentnih oblika epilepsije kod djece
Infantilni spazmi (IP) Kriteriji za dijagnosticiranje IP:
debi napadaje u prvoj godini života (maksimalno 4 - 9 mjeseci);
specifičnu prirodu napada u obliku kratkih fleksora, ekstenzora ili fleksora-ekstenzora kontrakcija mišića vrata, torza i ekstremiteta;
visoka učestalost napadaja tokom dana, serija;
odloženi psihomotorni razvoj različite težine;
specifična gigaritmija EEG uzorka (hipersinkronizirani ritam, prevlast sporih valova visoke amplitude, miješani s periodima difuznog brzog ritma, ili epizode poravnavanja krivulje);
otpornost na glavne antikonvulzive.
IP tretman
Osnovni lekovi u tretmanu IP su derivati valproične kiseline. Prosečna terapijska doza je 20–70 mg kg dnevno. Međutim, maksimalne doze koje smo koristili bile su 200 mg / kg dnevno. U zavisnosti od prirode napada (grčeva), kao dodatna terapija valproatu se propisuju sledeći lekovi: benzodiazepini, lamotrigin. sukcinimidi, karbamazepin. Značajno poboljšanje (smanjenje napada za 75-100%) postignuto je u 78% slučajeva. Sa apsolutnom otpornošću na ove lekove, moguće je sprovesti hormonsku terapiju (ACTH, sinakten depot, kortikotropin, prednizolon, deksazon) u kombinaciji sa antikonvulzantima.
Lennox Gasta sindrom
Kriteriji za dijagnosticiranje SLG (prema Lennox - Gastaut - Aicardi):
etiološka heterogenost;
debi napadaji u dobi od 1 7 godina;
polimorfizam epileptičnih napadaja kod jednog pacijenta: atipične izostanke, mioklonični napadi (čvorovi, pecks, trzanja), atono-astatički i toničko-astatski napadi, kratki tonički napadi, posebno u spavanju, kloničnim i toničko-kloničnim napadima, rjeđe parcijalni napadaji;
visoka učestalost napada tokom dana;
varijabilnost napadaja po danu (dobri i loši dani);
mentalna i govorna retardacija;
EEG uzorak-difuzna aktivnost sporog vrha je bilateralna i sinhrona sa frekvencijom od 1 2,5 Hz, obično s naglaskom na frontalne i temporalne režnjeve. Sleep (slow phase) oštro izaziva patološku aktivnost tipa polispike-vala sa frekvencijom od 10 Hz, što je karakteristično za tonične napadaje.
Tretman SLH
Osnovni lekovi su derivati valproične kiseline; Prosječna terapeutska doza je 30 100 mg na 1 kg tjelesne težine. Efikasne kombinacije, zavisno od prevalencije određenih napada, su: valproat + sukcinimid, valproat + lamotrigin, valproat + karbamazepin.
U prisustvu generaliziranih kloničkih (toničko-kloničnih) napada i statusnog tijeka napadaja, derivati barbiturne kiseline mogu se propisati kao treći lijek. Efikasnost tretmana je 70%.
Dijagnoza i liječenje idiopatskih oblika epilepsije
Idiopatski oblici epilepsije općenito se odnose na benigne oblike. Međutim, u nekim slučajevima, napadi su otporni na osnovne antikonvulzive. Naglašen je nedostatak terapijske efikasnosti u oblicima kao što su juvenilna apsantija - epilepsija, epilepsija sa miokloničkom apsantijom, epilepsija sa mioklono astatičkim napadima (ova dva oblika se često nazivaju kriptogenom generalizovanom epilepsijom).
Pedijatrijska apscesna epilepsija (DAE)
Kriteriji za dijagnozu DAE:
debi u 3 - 8 godina;
češće djevojke pate;
tipični kompleksni absans su glavni tip napadaja;
karakteriše ga najveća učestalost napada: desetine i stotine dnevno;
u oko 30% slučajeva moguć je pristup generaliziranih konvulzivnih napadaja;
tipičan EEG obrazac generalizirane aktivnosti vršnog vala sa frekvencijom od 3 Hz, koji se javlja, naročito često, s hiperventilacijom.
Principi tretmana DAE:
Osnovni preparati u odsustvu generaliziranih konvulzivnih napada sukcinimida i valproata; u prisustvu generaliziranih konvulzivnih napadaja isključivo valproaty. Prosječne terapijske doze sukcinimida su 10-15 mg / kg dnevno u 2 doze, za valproate 30-50 mg / kg dnevno. u 3 4 prijema. Rezervni lijekovi za benzodiazepin i lamotrigin. U rezistentnim slučajevima koriste se sledeće kombinacije: valproat + sukcinimidi; valproat + benzodiazepini; Valproatum Lamotrigine. Kompletna terapijska remisija među ispitanicima kod nas je postignuta u 70% slučajeva, a kod drugih je došlo do značajnog smanjenja učestalosti napadaja.
Juvenilna apsantija epilepsija (UAE)
Kriteriji za dijagnozu UAE:
debiji od 8 godina i stariji (maksimalno 9 13 godina);
jednostavna tipična apsancija (kraća i rjeđa nego kod DAE), glavni tip napadaja;
visok rizik od pridruživanja generaliziranih konvulzivnih napada do 75%;
za EEG je karakteristična pojava generalizirane vršne aktivnosti s frekvencijom od 4 Hz i više.
Principi tretmana UAE
Osnovni preparati isključivo izvedeni iz valproične kiseline. Prosečna terapeutska doza od 30 do 50 mg / kg dnevno u 3 do 4 doze. U rezistentnim slučajevima, posebno u prisustvu čestih generaliziranih konvulzivnih napadaja, moguće su kombinacije: valproat + barbiturati, valproat + lamotrigin. Potpuna terapijska remisija se postiže rjeđe nego kod AED, 56% slučajeva i značajno poboljšanje od 37%. Prognoza se pogoršava sa dodatkom čestih generaliziranih konvulzivnih napadaja.
Epilepsija sa izolovanim generaliziranim konvulzivnim napadima (GSP).
Kriteriji za dijagnozu SHG:
debi u vrlo širokom dobnom rasponu od 3 do 30 godina (u prosjeku 13-17 godina);
manifestuje se isključivo toničko-kloničkim konvulzivnim napadima, obično ograničenim na buđenje ili zaspanje;
učestalost napada je mala, rijetko prelazi 1 put mjesečno;
vremenom, moguće je dodati izostanke ili mioklonične napadaje sa transformacijom u odsutne oblike epilepsije ili juvenilne mioklonične epilepsije.
Principi tretmana za SHGs
Osnovni lek je karbamazepin. Prosječna doza je 15 do 25 mg / kg dnevno u 3 doze. Rezervne preparate valproata, barbiturata, hidantoina. U rezistentnim slučajevima moguće su kombinacije: karbamazepin + valproat; karbamazepin + barbiturati; karbamazepin + hidantoini: valproati + barbiturati; barbiturati + hidantoini. Pri vezivanju apsana ili miokloničnih napadaja, neophodna je hitna zamena karbamazepina valproatima. Potpuna remisija u 70% slučajeva i značajno smanjenje napada od 27%.
Epilepsija- hronična bolest mozak, koji se manifestuje ponovljenim neprovociranim napadima sa oštećenim motornim, senzornim, autonomnim, kognitivnim, mentalnim funkcijama uzrokovanim prekomernim neuronskim pražnjenjem u sivoj tvari moždane kore.
Prikazana definicija sadrži dvije važne odredbe: 1) samo ponovljeni napadi su osnova za uspostavljanje dijagnoze epilepsije; 2) epilepsija uključuje spontane, ne-provokativne napade (sa izuzetkom refleksnih oblika, na primjer, fotoosjetljiva epilepsija). Epilepsijski febrilni napadi, kao i napadaji koji se javljaju kod akutnih bolesti mozga (na primjer, encefalitis, subduralni hematom, akutna cerebralna cirkulacija, itd.) Nisu epilepsija.
Moderne ideje o bolesti počele su se pojavljivati tek krajem XIX vijeka. J. Jackson je 1888. godine definirao epilepsiju kao "... slučajno, iznenadno i prekomjerno lokalno uznemiravanje sive tvari u mozgu"; opisali su "insularne napade" (olfaktorne halucinacije sa temporalnom epilepsijom) i "stanja snova" (napadi sa oslabljenim mentalnim funkcijama). A.Y. Kozhevnikov (1898) je sve oblike epilepsije podelio na „organski“ (u modernoj terminologiji, simptomatske) i ustavne (idiopatske). Prvi pokušaj klasifikacije epileptičkih napadaja napravio je engleski neurolog V. Govers 1903. Sindromološki pristup u dijagnostici epilepsije uspostavio je V. Lennox 1961, H. Gastó 1966. i G. Doze 1980. godine. Ruski naučnici PM Sarajishvili i V.A. Karlov
Krajem XX veka. epilepsija je postala izlečiva bolest. Trenutna klasifikacija epileptičkih sindroma u 1989. godini kaže da postoje mnogi oblici epilepsije (sindromi) koji imaju vlastite obrasce razvoja i prognoze razvoja u zavisnosti od toga koja električna pražnjenja nastaju u moždanoj kori, gdje su lokalizovana, kako se šire i transformišu, i koji se napadaji kada se to dogodi kod pacijenta. U proučavanju epilepsije, važnu ulogu imaju metode neuro-snimanja (CT, MRI visoke rezolucije, PET, SPECT), digitalni EEG i video EEG monitoring. Trenutno, oko 65% slučajeva epilepsije je potpuno lečivo; u 20% slučajeva to se postiže kirurškim metodama.
Stav prema bolesnicima se promijenio, njihova socijalna adaptacija se poboljšala. Međutim, do sada nisu proučavani mnogi mehanizmi patogeneze ove teške bolesti; postoji veliki broj atipičnih oblika koji značajno ometaju tačnu dijagnozu; neki rezistentni oblici epilepsije i dalje ostaju nepovredivi.
Prevalencija epilepsije u opštoj populaciji iznosi 0,5-0,75%, au rasadniku 1%. Kod 75% pacijenata epilepsija debituje u djetinjstvu i adolescenciji, što je jedna od najčešćih patoloških stanja dječja neurologija.
Svi oblici epilepsije prema etiologiji podijeljeni su na idiopatske, simptomatske i kriptogene.
For idiopatske forme karakteriše ga normalna inteligencija, odsustvo fokalnih simptoma i strukturnih promena u mozgu pacijenta, kao i genetska predispozicija (slučajevi epilepsije kod rođaka). Etiologija je uglavnom uzrokovana kanalopatijama - genetski određenom difuznom nestabilnošću neuronskih membrana. Identifikovani su geni tri glavna monogeno nasleđena oblika epilepsije: autosomno dominantna frontalna epilepsija sa noćnim paroksizmima (loci 20ql3.2 i 15q24), benigni porodični grčevi novorođenčadi (loci 20ql3.2 i 8q24) i generalizirana epilepsija sa febrilnim obrascima i srčanom insuficijencijom. , mutacija gena SCN1B; 2q21-q33, mutacija gena SCN1A). Drugi oblici su određeni s nekoliko gena (poligensko nasljeđivanje). To uključuje adolescentsku miokloničnu epilepsiju, rolandičnu epilepsiju, benignu parcijalnu (porodičnu) epilepsiju djetinjstva, itd. Sa praktične tačke gledišta, treba imati na umu da ako jedan roditelj ima idiopatsku epilepsiju, vjerovatnoća da ima bolesno dijete nije veća od 10%.
Simptomatski oblici epilepsiju karakteriše obavezno prisustvo morfološkog supstrata: tumori, ciste, glijalni ožiljci, abnormalnosti mozga i aneurizme. Oni se detektuju pomoću tehnika neuro-snimanja.
Term "Cryptogenic" (“Pretpostavlja se da je simptomatska geneza”) definira one oblike epilepsije, čiji uzrok ostaje nejasan čak i uz upotrebu svih modernih metoda istraživanja. Na primjer, u slučaju kombinacije epilepsije s hemiparezom ili kongenitalnom mentalnom retardacijom, pretpostavlja se simptomatska priroda bolesti, ali u CTI MRI-a nema nikakvih promjena u mozgu.
Focal napadi i oblici epilepsije objašnjeni su konceptom kortikalnog "epileptogenog fokusa", koji igra ulogu "pejsmejkera". Hipersinkroni iscjedak koji nastaje u njemu uključuje veliki broj neurona u korteksu, koji se šire u susjedna područja mozga.
Sa generalizovano napadi epilepsije generalizovani su od samog početka, što dokazuju EEG podaci (bilateralna sinhrona distribucija u obje hemisfere). Patogeneza generalizovanih oblika epilepsije još uvijek nije dovoljno jasna. Vodeća thalamo-kortikalna hipoteza objašnjava pojavu primarne generalizacije integrativnim sistemom koji se sastoji od cerebralnog korteksa i talamusa (thalamo-cortical i cortico-thalamic pathways). Izvor pražnjenja se verovatno nalazi u cerebralnom korteksu, thalamo-kortikalne veze sinhronizuju generalizovana pražnjenja, a retikularna formacija debla (prvenstveno srednji mozak) modulira nivo „preosetljivosti“ korteksa na pražnjenja. U distribuciji i generalizaciji epileptičkog iscjetka učestvuju i cingularni girus, orbito-frontalni korteks, amigdalo-hipokampalni kompleks, substantia nigra. Tokom stimulacije talamokortikalnih sistema na EEG, može se javiti generalizovana aktivnost vršnih talasa, kao i bilateralna sinhrona paroksizmalna pražnjenja ritmičkih delta talasa.
Primarna generalizirana epilepsija javlja se u stanju abnormalno visoke ekscitabilnosti talamikokortikalnog sistema. Nivo ekscitabilnosti je verovatno određen genetski i zbog nestabilnosti membrana neurona i nemogućnosti održavanja normalnog gradijenta jona Na, K i Cl.
Klasifikacija epileptičkih napadaja usvojila ga je Međunarodna liga protiv epilepsije 1981. godine u Kjotu (Japan). Epileptički napadi se dijele na: 1) žarišne (žarišne, žarišne, lokalne, lokalno uzrokovane); 2) uopšteno; 3) nisu klasificirani (tabela 20).
Fokalni (žarišni, fokalni) napadi dijagnostikovan u slučaju kada na početku paroksizma postoje jasni klinički i elektrofiziološki kriteriji za uključivanje određenih moždanih struktura. Na primjer, u slučaju kloničnih konvulzija, pola lica i ruke na jednoj strani (faciobrachial napadaji), epileptički fokus se nalazi u srednjim dijelovima prednjeg dijela.
central gyrus; sa olfaktornim halucinacijama - u području kuke temporalne giruse; na fotoskopijama - u korteksu potiljnog režnja; sa "neuspjehom misli" (dismneički napadi) - u frontalnom režnju, itd. Sa jednostavnim parcijalnim napadima, svijest nije poremećena. Na EEG-u tokom napada počinje lokalni epileptički iscjedak, počevši od odgovarajućeg područja moždane kore.
Fokalni napad sa sekundarnom generalizacijom može početi kao parcijalni, ali zatim prelazi u generalizovano, uključujući sve mišiće trupa i ekstremiteta, sa širenjem epileptiformne aktivnosti na EEG na obje hemisfere.
Teški fokusni napadi nastavite sa oslabljenom svešću (tokom napada pacijent ne reaguje na adresirani govor, ne izvršava komande, amnezira napad). EEG tokom kompleksnog parcijalnog napadaja otkriva pojedinačni ili bilateralni epileptički iscjedak, obično u temporalnim ili frontalnim elektrodama (Tabela 21).
To generalizirani napadaji uključuju tipične i atipične izostanke, klonične, toničke, kloničko-tonske i atonske napade, kao i miokloniju.
Tabela 20Međunarodna klasifikacija epileptičkih napadaja (Kyoto, 1981)

Utvrđeno je da epilepsija nije pojedinačna bolest sa različitim napadima, i podeljena je na odvojene oblike -
epileptički sindromi. Odlikuje ih stabilan odnos kliničkih, električnih i anatomskih kriterija; razlikuju se u odgovoru na antiepileptičku terapiju i prognozu (Tabela 21).
Tabela 21.Promene u EEG-u sa različitim napadima
Tabela 22.Međunarodna klasifikacija epilepsije, epileptičkih sindroma (New Delhi, 1989)
1. Oblici epilepsije povezani sa lokalizacijom (žarišna, lokalna, fokalna)
1.1. Idiopatski (sa nastupom starosti)
Benigna epilepsija djetinjstva sa vrhovima centralnog vrha (rolandski).
Epilepsija djetinjstva s okcipitalnim paroksizmom.
Čitanje primarne epilepsije.
1.2. Simptomatski
Hronična progresivna parcijalna epilepsija (Koževnikov sindrom).
Napadi, karakterizirani specifičnim metodama provokacije.
Ostali oblici epilepsije sa poznatom etiologijom ili organskim promjenama u mozgu.
1.3. Cryptogenic



Treba napomenuti da je u prošlosti od 1989. godine nesavršenost klasifikacije postala očigledna, jer nije uključivala neke oblike (npr. Pseudolenoks sindrom). Pored toga, mnogi simptomatski oblici Westovog sindroma i Lennox-Gastautovog sindroma ne odnose se na generaliziranu epilepsiju, jer predstavljaju parcijalnu epilepsiju sa fenomenom sekundarne bilateralne sinhronizacije. Godine 2001. Međunarodna komisija za klasifikaciju i terminologiju izdala je nacrt nove klasifikacije epileptičkih napadaja i epileptičkih sindroma (Tabela 22). Pored klasične podjele na fokalne i generalizirane napadaje, to ukazuje da u odnosu na mnoge benigne i samostalne epileptičke sindrome, termin "epilepsija" treba zamijeniti sa "napadajima". Na primjer, ne „alkoholna epilepsija“, već „napadaji povezani s ukidanjem alkohola“ itd. Mnogi novi oblici epilepsije opisani su kao jasno utvrđeni, uvedeni su novi termini. Termin "parcijalni napadi i parcijalna epilepsija" zamjenjuje se "fokalnim napadima i fokalnom epilepsijom"; "Kriptogene forme" do "verovatno simptomatske forme." U definiciji sindroma preporučuje se zamjena riječi "napadaji" sa "napadajima". Koncept “napadaja” je mnogo širi od pojma “napadaji”, a daleko od svih napadaja manifestiraju se upravo napadima. Eliminisana je podela fokalnih napada na jednostavne i složene u zavisnosti od poremećaja svesti, jer u većini slučajeva procena nivoa svesti ostaje indikativna. Prednost klasifikacije je razvoj koncepta dječje epileptične encefalopatije.
Dijagnostikaepilepsija uključuje sljedeći algoritam:
1. Opis paroksizmalnih događaja (možda samo prema anamnezi).
2. Klasifikacija napadaja (istorija, klinika, EEG, video EEG monitoring).
3. Dijagnoza oblika (povijest, klinika, EEG, video EEG monitoring, neuroimaging).
4. Uspostavljanje etiologije (MRI, kariotipiranje, biokemijsko istraživanje, biopsija mišića, itd.).
5. Dijagnoza popratnih bolesti i utvrđivanje stepena invalidnosti.
Dijagnoza epilepsije je klinički-elektro-anatomski. U XXI veku. da bi se ustanovila tačna dijagnoza epilepsije, nije dovoljno imati opis napada koje su predstavili rođaci. Neophodna je elektroencefalografska potvrda (električni kriterij), kao i tehnike snimanja (anatomski kriterij). Za tačno određivanje dijagnoze i propisivanje pravilne terapije, osim rutinskih metoda, potrebno je sprovesti dugoročni EEG video nadzor, noćni EEG monitoring, MRI visoke rezolucije u 3D modu vizuelizacije itd.
14.1. Idiopatske fokalne forme
Benigna parcijalna epilepsija detinjstva sa centralnim temporalnim vrhovima (rolandska epilepsija) [ER] - karakterišu kratki pharyngooral i hemifacial motorički napadaji koji se obično javljaju kod buđenja i pada u snu, kao i tipične promjene u EEG-u (slika 14.1). RE je najčešći oblik epilepsije u djetinjstvu. Stopa incidencije je 21 na 100.000 dječje populacije.
Bolest počinje u dobi od 2 do 14 godina (maksimalno 7-9 godina), dječaci su češće bolesni. Karakterizirani su jednostavnim fokalnim napadima koji se javljaju u 80% slučajeva kod buđenja ili zaspanja. Napad počinje sa somatosenzornom aurom: trnci, obamrlost na jednoj strani ždrela, jezik, desni. Tada pacijenti prave osebujan grkljan zvuk kao što je "grgljanje", "grunting", "gargling"; uočena je hipersalivacija i anarthria (faringeoralni napadi). Karakteristične konvulzije mimičkih mišića: jednostrani tonik, klon
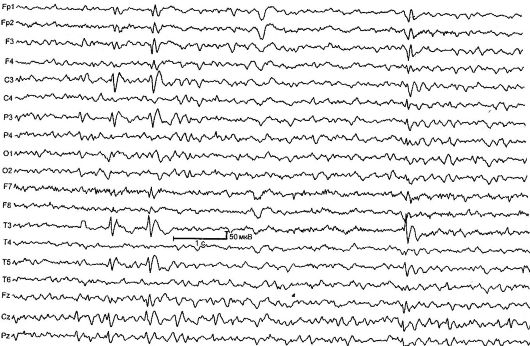
Sl. 14.1.EEG dijete star 4 godine sa rolandičnom epilepsijom
ili toničko-klonične konvulzije mišića lica, usana, kao i jezika, ždrijela, larinksa (hemifacijalni napadi). Kod 20% pacijenata, konvulzije su se proširile od mišića lica do homolateralne ruke (brahiofacijalni napadaji); u oko 8% slučajeva se pojavljuju iu nozi (unilateralni napadaji). Kako bolest napreduje, napadaji mogu promijeniti stranu.
Sekundarni generalizirani konvulzivni napadaji javljaju se kod 25% djece. Napadi na OM traju od nekoliko sekundi do 1-2 minuta. Učestalost njihovog prosjeka - 2-6 puta godišnje. Vremenom se javljaju sve manje i manje (čak i bez tretmana) i ne primećuju se kod odraslih.
Promene na EEG-u u interiktalnom periodu utvrđene su u 90% slučajeva, tipičan uzorak je kompleks akutnog sporog talasa. Početna komponenta obično se sastoji od trofaznog akutnog talasa praćenog sporim talasom, što stvara sličnost sa kompleksima QRSTna EKG-u. Ova aktivnost je lokalizovana u centralno-temporalnim vodama i naziva se "rolandska" ili ima opće ime - "benigni epileptiformni poremećaji djetinjstva" (DEND). Da bi se potvrdila dijagnoza, važno je voditi
EEG tokom noćno-noćnog EEG monitoringa, kao i kod oko 30% dece sa RE, rolandski kompleksi se detektuju samo tokom sna.
TerapijaUzimajući u obzir benigni tok, moguće je ne odrediti anti-epileptičku terapiju. Međutim, nije isključena dijagnostička greška, kao i mogućnost pretvaranja ree u recesiju u pseudo-linox sindromu u približno 5% slučajeva kod djece mlađe od 7 godina. Preporučuje se početak terapije ponovljenim napadima. Liječenje se uvijek provodi s jednim lijekom (polieterapija je neprihvatljiva), počevši s derivatima valproinske kiseline (depakin, konvuleks, konvulsofin). Valproati se propisuju postepenim povećanjem doze do 15-30 mg / kg dnevno (u proseku 600-1500 mg / dan) u 2 doze.
Uz neefikasnost ili netoleranciju valproata, topiramat (topamax) se propisuje u dozi od 50-150 mg / dan (3-5 mg / kg). Također se koriste lijekovi iz skupine karbamazepina (Tegretol, Finlepsin) u prosječnoj dnevnoj dozi od 15-20 mg / kg (300-600 mg / dan). U nekim slučajevima, karbamazepin može dovesti do povećanja DEND indeksa na EEG i povećanja napadaja - fenomena pogoršanja. U tom smislu, ne preporučuje se propisivanje karbamazepina kao početne terapije, kao ni u svim slučajevima kod djece mlađe od 7 godina. Upotreba barbiturata i hidantoina je kontraindikovana!
Potrebno je praćenje EEG-a, uključujući praćenje EEG spavanja. Remisija u EM se postiže u 100% slučajeva do dobi od 16 godina.
Idiopatska parcijalna epilepsija sa okcipitalnim paroksizmima (benigna zatiljna epilepsija, DZE)- karakteriše ih napadi sa oštećenim vizuelnim funkcijama, simptomi slični migreni i prisustvo DEND obrasca na EEG-u u okcipitalnom području. ECD čini oko 20% svih idiopatskih parcijalnih oblika dječje epilepsije. Identifikovane su dve varijante DEC-a: sa ranom i kasnom manifestacijom bolesti.
Benigna epilepsija sa ranim debutom (Panayotopoulos sindrom) počinje između 1 i 13 godina, sa vrhuncem manifestacije od 3-6 godina. Bolest se manifestuje u retkim teškim napadima sa vegetativnim poremećajima, produženim gubitkom svesti i tendencijom ka statusnom kursu. Napadi se javljaju u snu, posebno prije buđenja; početi sa povraćanjem, glavoboljom, blanširanjem lica, nakon čega slijedi okretanje glave i očiju u stranu. Napadi obično završavaju hemikonvulzivnim ili generaliziranim napadima. Pojavljuje se "iktalna sinkopa", koja se manifestuje produženim
gubitak svijesti i nagli pad mišićnog tonusa, traje od 30 minuta do 7 sati, u prosjeku 2 sata, a većina pacijenata ulazi u jedinicu intenzivne njege. "Iktalna sinkopa" može da prethodi sekundarno-generalizovanim toničko-kloničnim napadima ili da se odvija odvojeno od njih. Uprkos teškom kursu statusa, učestalost takvih napada je mala. U nekim slučajevima postoji samo jedan napad za čitavo razdoblje bolesti. Prognoza je apsolutno povoljna.
Kasni debi benigni okcipitalni epilepsija (Gastho oblik) debituje od 3 do 15 godina, u prosjeku 8 godina. Karakteristični su jednostavni fokalni senzorni napadi sa oštećenjima vida u obliku jednostavnih vizualnih halucinacija (male višebojne kružne figure), koje se često javljaju u perifernom vidnom polju i kreću se u suprotnom smjeru od centra. Napadi traju od nekoliko sekundi do 1-3 minuta. Halucinacije se mogu pojaviti u istoj polovini vidnog polja. Raznovrsna komponenta se često primjećuje - okretanje očiju i glave je kontralateralno do ognjišta, dok um ostaje netaknut. Napadi mogu rezultirati unilateralnim ili sekundarnim generalizovanim toničko-kloničnim napadima. U polovini pacijenata nakon napada javlja se intenzivna pulsirajuća migrena kao glavobolja, praćena mučninom i povraćanjem. Učestalost napada je obično mala, iako u nekim slučajevima mogu biti nedjeljni. Na EEG-u se detektuju visoko-amplitudni akutno-spori valni kompleksi koji se javljaju kod 2/3 pacijenata samo u okcipitalnim vodovima. Morfologija kompleksa je slična benignim epileptiformnim poremećajima u detinjstvu. U 1/3 pacijenata, epileptiformna aktivnost može se zabilježiti u drugim područjima (češće u centralno-temporalnim tragovima).
TerapijaLijekovi prvog izbora u liječenju DZE su soli valproične kiseline (depakin, konvulex, konvulsofin) u prosječnoj dnevnoj dozi od 30-40 mg / kg. Lijek se propisuje u dvije doze s maksimalnom dozom u večernjim satima.
Uz nedovoljnu efikasnost, moguća je monoterapija karbamazepinom (finlepsin, tegretol) u prosečnoj dozi od 15-20 mg / kg / dan ili topiramat u dozi od 75-200 mg / dan (3-6 mg / kg / dan).
Kod Panayotopulos sindroma, kompletna remisija napada za 9 godina javlja se kod 92% pacijenata. Kod pacijenata sa oblikom Gastoma, remisija se uočava u 82% slučajeva do dobi od 15, au 100% do 18 godina.
Autosomno dominantna frontalna epilepsija sa noćnim napadima
je idiopatski oblik. Identifikovani lokus gena odgovoran za razvoj ove bolesti: 20q13.2i 15q, ali ima i sporadičnih slučajeva. Debitantsko doba varira od 2 meseca do 52 godine, sa maksimumom u prvoj deceniji života. Konvulzije kod 70% pacijenata počinju sa nespecifičnom aurom: "hladnoća tremora", glavobolja, slušnih halucinacija, vrtoglavice, somatosenzornih senzacija (svrbež torza), nakon čega su tipični napadi sa hipermotomatskim automatizmima. Počinju sa konvulzivnim disanjem, gruncanjem, jakim krikom zavijanja. Oči širom otvorene, izraz užasa na licu. Pacijent pogleda gore, sjedne u krevet; pojavljuju se hipermotori i distoni fenomeni. Ponekad pacijent (obično odrasla osoba) pravi haotične pokrete rukama (poput boksačkih pokreta) i nogama (poput pedaliranja); ustaje na sve četiri i pravi zamah pokretima sa karlicom. Svest tokom napada obično nije poremećena. Karakterizira se pojavom napada isključivo u snu, oni se mogu ponavljati više puta preko noći kao serija, onda postoji pauza za nekoliko dana ili sedmica i serija se ponovo nastavlja. Trajanje napada - od nekoliko sekundi do 1 min. U rijetkim slučajevima moguće je pojavljivanje sekundarnih generaliziranih paroksizama.
EEG budnosti nije specifičan. Podaci EEG monitoringa noćnog sna i video EEG monitoringa su dijagnostički značajni, koji otkrivaju nisko-amplitudnu epileptiformnu aktivnost u obliku akutno sporog talasnog kompleksa, koji se javlja regionalno u jednom od frontalnih, frontalno-temporalnih vodova ili bifrontalno asinhrono.
Početak liječenja počinje lijekovima karbamazepin, dva puta s maksimumom prije noćnog sna. Dnevna doza - 600-1000 mg / dan (15-30 mg / kg / dan). U slučaju neefikasnosti, topiramat se daje u dozi od 100-400 mg / dan (3-10 mg / kg / dan), dva puta s maksimumom prije spavanja. Sljedeća faza liječenja je monoterapija valproatom. Convulex se daje dva puta po dozi.
900-1800 mg / dan (20-40 mg / kg / dan).
U rijetkim slučajevima rezistencije može se primijeniti polieterapija, koja se sastoji od kombinacije dva osnovna AED-a (valproična kiselina s karbamazepinom ili topiramatom). U većini slučajeva postiže se remisija lijekova.
14.2. Simptomatska fokalna epilepsija
Simptomatska frontalna epilepsija (SLE) je lokalno uslovljena forma sa potvrđenim morfološkim poremećajima unutar frontalnih režnjeva velikog mozga. On čini 30-40% među svim simptomatskim fokalnim oblicima epilepsije i zauzima 2. mjesto po učestalosti nakon temporalne epilepsije (u djetinjstvu može nadmašiti temporalnu epilepsiju u smislu učestalosti).
Etiologija uključuje traumatske povrede mozga, tumore i ciste frontalnog režnja, fokalnu kortikalnu displaziju, gliozu kao rezultat perinatalne encefalopatije, vaskularne anomalije.
U okviru SLE razlikuje se nekoliko oblika.
Motor (premotor, Jackson) SLE javlja se kada je nadražena prednja središnja gyrus. Karakteristične su jednostavne fokalne motoričke napade sa konvulzijama u kontralateralnom nidusu. "Jackson" marš počinje grčevima ruke ili stopala, uz postepeno uključivanje ruku, nogu i mišića lica iste strane. Često se napad završava prolaznom Toddovom parezom.
Operativni SLE javlja se tokom stimulacije operularne zone frontalnog režnja. Odlikuje se kompleksnim fokalnim (dijaleptičkim) napadima s oro-alimentarnim automatizmima; ipsilateralno trzanje mišića lica, mogući su autonomni fenomeni.
Orbitofrontal SLE nastaje kada je orbitalni korteks iritiran donjim frontalnim gyrusom. Karakterišu ga kompleksni fokalni, autonomni-visceralni napadi, paroksizmi sa nasilnom vokalizacijom i atipične izostanke.
Dorsolateralni (prefrontalni) SLE nastaje iz zadnjeg dijela gornjeg i donjeg prednjeg gira. Ona se manifestuje toničnim neželjenim napadima sa okretanjem očiju i glavom u suprotnom smjeru od ognjišta; Takođe je moguće oteti i podići ruku, na koju je pacijent gledao. Često se javlja motorna afazija sa lokalizacijom fokusa u dominantnoj hemisferi.
Prednji polarni SLE nastaje kada je epileptogeni fokus lokaliziran u polnoj regiji frontalnih režnjeva. Predstavljena je jednostavnim parcijalnim napadima sa oslabljenim kognitivnim funkcijama (priliv misli, "neuspeh" misli, promena tokom vremena) i kompleksni parcijalni (dijalektički) napadi.
Tsingulyarnaya SLEopaženo uz iritaciju prednjeg cingularnog girusa. Ona se manifestuje kompleksnim parcijalnim napadima sa gesturalnim automatizmom, ipsilateralnim treptajućim pokretima, kao i “limbičkim paroksizmima”: izrazom straha, crvenilom lica, narušavanjem emocionalne sfere - disforijom.
SLE koji dolazi iz dodatnog motornog područja (premotor SLE), - jedan od najčešćih oblika frontalne epilepsije, karakteriziran kratkim posturalnim asimetričnim toničnim epizodama (spazama) koje se bilateralno javljaju u proksimalnim ekstremitetima (na primjer, kao što je "držanje mačevaoca"). Napadi uglavnom noćni, javljaju se serijski. Takođe su uočeni napadi govora koji se zaustavljaju sa jasnim umom ili vokalizacijom u obliku krikova, zavijanja. Mogući su napadaji sa stereotipnim hipermotor automatizmom: haotični pokreti sa rukama (prema tipu boksa), noge (pokreti pedaliranja) i karlica.
Napadi su kratki, sa kratkom ili nepotpunom dezaktivacijom svijesti, minimalnom konfliktnom postiktalnom reakcijom, serijskim ciklusolekularnim tokom i dominantnom pojavom noću.
Rezultati neurološkog pregleda zavise od etiologije SLE. U slučaju ekstenzivnih oštećenja frontalnog režnja (npr. Glomazna masa), hemipareza se otkriva na suprotnoj strani od fokusa (visoki refleksi, patološki refleksi); hemiaxia je moguća. Često se formira kršenje ponašanja tipa "frontalne psihe".
EEG u interiktalnom periodu je neinformativan ili nespecifičan. Dugoročni EEG monitoring je poželjniji (i nužno za vrijeme spavanja), koji otkriva regionalne epileptiformne obrasce (akutno-spor val), nastavak regionalnog usporavanja u jednom od frontalnih potencijala, fenomen sekundarne bilateralne sinkronizacije.
MRI se izvodi kako bi se otkrio strukturni defekt.
Početni tretman počinje topiramatom (topamax) u početnoj dozi od 12,5-25 mg / dan. Doza se postepeno povećava za 12,5-25 mg 1 put nedeljno do 50-500 mg / dan (3-10 mg / kg / dan), u 2 doze (ujutro i uveče) u intervalu od 12 sati. karbamazepin, koji se koristi u dozi od 600-1800 mg / dan (15-35 mg / kg / dan), 2 puta dnevno. Karbamazepin i okskarbazepin su posebno efikasni u dijabetičkim napadima. Sa "pseudo-generalizovanim"
prepone i fenomen sekundarne bilateralne sinhronizacije na EEG karbamazepinu kontraindiciran je jer može pogoršati napade.
Sredstva trećeg izbora - preparati valproične kiseline (konvulex, depakin, konvulsofin) koriste se u dozi od 1000-3000 mg / dan (30-60 mg / kg / dan), 2 puta dnevno.
Uz neefikasnost tri osnovna lijeka, preporuča se polieterapija - kombinacija topiramata ili valproata sa sukcinimidima. Etosuksimid (suksilep) se propisuje u dozama od 500-1000 mg / dan (20-40 mg / kg / dan) u 3 doze. U drugim slučajevima, propisati kombinaciju osnovnog AEP: topiramat + valproat, valproat + karbamazepin, karbamazepin + topiramat.
Rezervni lijekovi za polieterapiju su lamotrigin (lamictal) i levetiracetam (keppra). Lamotrigin (3-7 mg / kg / dan) se koristi samo u kombinaciji sa osnovnom AED. Prosečna doza je 100-400 mg / dan u kombinaciji sa topiramatom ili karbamazepinom i 100-200 mg / dan sa valproatom. Levetiracetam je efikasan u kombinaciji sa osnovnim AEP-om u dozi od 1000–4000 mg / dan (30–60 mg / kg / dan) za fokalne motorne i sekundarno generalizirane napadaje.
Prognoza bolesti sa SLE je uvek ozbiljna, što je povezano sa postojanjem strukturnog defekta u korteksu, hemiparezi i izraženom kognitivnom oštećenju. Remisija leka se postiže samo u 20% pacijenata. U drugim slučajevima, moguće je značajno smanjiti učestalost napada. Za otporne napadaje koristi se hirurški tretman. Glavni tip hirurške intervencije je fokalna kortikalna resekcija.
Simptomatska epilepsija temporalnog režnja (BOO) je lokalno uslovljena forma sa poznatom etiologijom i morfološkim poremećajima u temporalnim režnjevima mozga (amonijačna skleroza, benigni urođeni tumori temporalnog režnja, fokalna kortikalna displazija, posljedica perinatalnog oštećenja). Postoje dvije glavne forme ONA: limbic (sinonimi: paleokortikalna, amigdalo-hipokampalna) i nonokortikalna (sinonim: lateralna).
U 75% slučajeva počinju napadi auras.Koncept aure treba jasno definirati i razlikovati od prekursora epileptičkog napadaja. Pod arom (iz grčkog. - dah) treba razumjeti kliničke fenomene koji nastaju sami od sebe
ili prije sekundarnog generaliziranog ili djelomičnog napadaja. Aura je uzrokovana lokalnim epileptičkim pražnjenjem u određenom dijelu moždane kore i u suštini je jednostavni parcijalni napad. Priroda aure ukazuje na lokalizaciju ognjišta. Izdvajaju se sljedeće vrste aure: somatosenzorni, vizualni, olfaktorni, okusni, slušni, vrtoglavi, mentalni, vegetativni, abdominalni (abdominalni). Forerunnersjavljaju se mnogo minuta, sati ili dana prije epileptičkog napada, obično se manifestiraju mentalni ili autonomni simptomi koji nisu praćeni lokalnim kortikalnim pražnjenjima.
Amigdala-hipokampal (paleokortikalna, limbička) - najčešći oblik je oko 65% svih slučajeva SHE. Osnova bolesti često leži skleroza (glioza) mediobazalnih podjela temporalnog režnja usled perinatalnog oštećenja ili atipičnih febrilnih napadaja. Bolest obično počinje dugim, često hemiclonskim, febrilnim napadima mlađim od 3 godine. Zatim slijedi period imaginarnog blagostanja - nema napada sve do predpubertetskog perioda. Najtipičniji (70% slučajeva) kompleksni fokalni napadaji sa deaktivacijom svijesti (dijaleptički) ili automatizmom (automotor). Kod dijabetičkih napada, pacijent iznenada zaustavlja fizičku aktivnost, zamrzava se sa širokim očima, pogled izražava zaprepaštenje ili strah ("zureći pogled").
Automatizacija u obliku gestova (trljanje ruku, prsti, stiskanje ruke, miješanje odjeće) i oro-alimentarnih akcija (smacking, gutanje, lizanje) karakteristične su za BOOS. Automatizmi u četkici su uočeni na strani ognjišta, a distonična ugradnja prstiju ruke je na suprotnoj strani. Trajanje automatskih napada od 30 sekundi do 3 minuta, brzo postaju češći i postaju otporni na terapiju.
Često napadaji prate povrede vegetativnih funkcija. Posebno karakteristični epigastrični paroksizmi sa jasnom sviješću. Pacijent oseća bol, napetost, nelagodnost u pupku; moguće ispuštanje gasova. Ovaj „uzlazni epileptički osjećaj“ se diže od trbuha do grla, praćen osjećajem stiskanja vrata, nakon čega je moguće isključiti svijest.
Odlikuje se i jednostavnim fokalnim napadima sa oštećenim mentalnim funkcijama: Jackson-ova stanja snova ("sanjalačka stanja"), koja se manifestuju iznenadnim osećanjima
"Snovi budni"; osjećaj "već viđenog" ili "nikad viđenog"; nastanak derealizacije (osećaj nestvarnosti okoline) ili depersonalizacija (narušavanje samo-percepcije). Uključivanjem kompleksa u obliku badema pojavljuju se kratki napadi nemotiviranog straha, disforije, agresije.
Lateralna (neokortikalna) ONA javlja se pri lezijama gornjih lateralnih dijelova temporalnog režnja. Mogući su sledeći tipovi napada: slušne halucinacije (paroksizmalni osećaji buke, muzike, glasovi); vizuelne halucinacije (paroksizmalni izgled složenih svijetlih panoramskih vizualnih slika, često sa elementima sjećanja na prošle događaje); napadi nesistemske vrtoglavice, često u kombinaciji s vegetativnim manifestacijama (bljedilo kože, hiperhidroza, tahikardija); paroksizmalna senzorna afazija sa lokalizacijom epileptičkog fokusa u dominantnoj hemisferi; "Vremenska sinkopa" sa gašenjem svijesti, šepanjem i polaganim padom bez napada.
Neurološki pregled često otkriva piramidalne simptome kontralateralnog fokusa: oslabljeni VII i XII kranijalni nervi, asimetrija mišićnog tonusa, anisorefleksija i patološki refleksi. Odrasli pacijenti sa dugotrajnim tokom bolesti razvijaju ličnost i kognitivna oštećenja, označena terminom "glissroid": viskoznost, ukočenost, inercija mišljenja, poteškoće pri promeni, "lepljenje" na sitnice, upornost afekta; smanjena memorija i pažnja.
EEG u interiktalnom periodu u 50% slučajeva - bez patoloških promjena. Aktivnost vršnog vala u temporalnim režnjevima je zabilježena u ne više od 20% bolesnika.
MRI u koronarnoj projekciji može otkriti hipokampalnu sklerozu, ekspanziju donjeg roga lateralne komore, smanjenje volumena zahvaćenog temporalnog režnja, u nekim slučajevima - fokalnu kortikalnu displaziju.
Liječenje započinje lijekovima karbamazepina (finlepsin retard, tegretol CR), u dozi od 600-1800 mg / dan (15-35 mg / kg / dan) u 2 doze u 12-satnom intervalu ili u 3 doze sa 8-satnim intervalom. Okskarbazepin (trileptal) se propisuje u dozi od 600-2400 mg / dan (20-40 mg / kg / dan). Lijek drugog izbora - topiramat, propisan je, postepeno povećavajući dozu na 100-400 mg / dan (4-8 mg / kg / dan), 2 puta dnevno.
Sredstva trećeg izbora - lekovi valproične kiseline koriste se u dozi od 1000-3000 mg / dan (30-70 mg / kg / dan) u 2 ili 3 doze u jednakim vremenskim intervalima.
Uz neučinkovitost tri osnovna lijeka, preporuča se polieterapija: kombinacije karbamazepina (ili okskarbazepina) s valproatima, topiramatom; valproat sa topiramatom. Rezervni lijekovi za polieterapiju - lamotrigin (3-7 mg / kg / dan, samo u kombinaciji sa osnovnom AED) i levetiracetam.
F rognosis.Remisija leka se postiže samo kod 1/3 pacijenata. Kod preostalih pacijenata u većini slučajeva moguće je značajno smanjiti učestalost napada. U slučajevima otpornim na lekove primenjuje se hirurško lečenje, posebno selektivna amigdalo-hipokampotomija.
Simptomatska okcipitalna epilepsija (SZE) karakteriše prisustvo epileptogenog fokusa i morfološke promene u okcipitalnom regionu. Etiološki faktori su fokalna kortikalna displazija, posljedica perinatalnih lezija, okcipitalna kalcifikacija celijakije, vaskularne anomalije (Sturge-Weberov sindrom), MELAS, progresivna mioklonusna epilepsija s Laurus telima tijela, tumori, ONMK u stražnjoj cerebralnoj arteriji.
Dob početka SZE je promenljiva. Utvrđene su sledeće vrste napada: jednostavni fokalni senzor sa vidnim poremećajima (makro-, mikropsija, elementarne vizuelne halucinacije), sa okulomotornim poremećajima (adverzija glave i oka na suprotnu stranu, prisilni paroksizmalni treptaj, nistagmus); vegetativno-visceralno (mučnina, povraćanje, glavobolja); sekundarna generalizovana konvulzija. Često se u strukturi napada uočavaju amauroza i homonimni kvadrant hemianopija (ili kao simptomi prolapsa nakon napada). Karakteristična je posthumna migrena kao glavobolja.
U nekim slučajevima, neurološki pregled je određen strabizmom, ambliopijom, suženjem vidnih polja ili hemianopije. EEG studija u interiktalnom periodu kod 30% bolesnika sa SZE ne otkriva patološke promjene. Češće se određuje regionalno usporavanje ili epileptiformna aktivnost vršnih talasa u jednom od okcipitalnih vodova ili bioccipitalno sa prevlastom amplitude na strani fokusa.
Neurovizualizacija otkriva okcipitalnu kortikalnu displaziju, lokalnu gliozu zbog perinatalne encefalopatije (umora), kalcifikacije, vaskularne anomalije.
Tretmanpočnite sa preparatima karbamazepina u dozi od 600–1800 mg / dan (15–35 mg / kg / dan), u 2 doze, sa intervalom od 12 sati. Visoke doze karbamazepina su posebno efikasne za izolovane vizuelne aure i fokalne napadaje sa oštećenim vegetativnim funkcijama. Mnogi autori preporučuju započinjanje liječenja SZE okskarbazepinom u dozi od 600-2400 mg / dan (20-40 mg / ksut).
Lijek drugog izbora, topiramat, propisuje se u dozi od 100-400 mg / dan (5-8 mg / kg / dan) 2 puta dnevno. Sa sekundarnom bilateralnom sinhronizacijom na EEG, Topamax može biti starter.
Lijek trećeg izbora je valproična kiselina. Prosječna doza je 1000-2000 mg / dan (30-60 mg / kg / dan), ako je potrebno, veća, u 2 ili 3 doze.
U rezistentnim slučajevima koristi se polieterapija. Kombinacije karbamazepina (ili okskarbazepina) sa valproatima, valproatima sa topiramatom i ređe karbamazepinom sa topiramatom su posebno efikasne. Prilikom dodavanja drugog leka, doza prvog, po pravilu, ne opada. Rezervni lijekovi za polieterapiju - lamotrigin i levetiracetam.
Prognozazavisi od prirode strukturnog defekta mozga i načina širenja ekscitacije u korteksu. U 40-50% bolesnika može se postići trajna remisija lijekova. U rezistentnim slučajevima SZE u odsustvu efekta primene AED, jedini metod prave brige o pacijentima je neurohirurška intervencija - kortikalna resekcija.
Kozhevnikov epilepsija i Rasmussenov encefalitis (EC) je polietiološka bolest, koja se manifestuje kombinacijom mioklonskih, fokalnih motornih, sekundarno-generaliziranih napada sa fokalnim neurološkim simptomima.
Bolest je prvi opisao ruski neurolog profesor Aleksej Jakovljević Kozhevnikov pod nazivom "epilepsia corticalis sive partialis continua". 21. januara 1894. godine, na sastanku Moskovskog društva neurologa i psihijatara koje je on stvorio, on je napravio prezentaciju na temu "O posebnoj vrsti kortikalne epilepsije". Izvještaj je zasnovan na studiji o 4 slučaja kortikalne epilepsije, koju je autor primijetio u moskovskoj klinici za nervna oboljenja, i bio je
originalni opis bolesti, do tada još nije poznat. Klinička slika bolesti kod svih 4 bolesnika bila je izuzetno slična: “... kombinacija generaliziranih epileptičkih napadaja sa trajnim kloničnim konvulzijama u strogo određenim dijelovima tijela. Od ovih upornih napadaja bilo je: 1) tipični Jackson napadi u jednoj polovini tijela, i 2) gore spomenuti opći napadi, koji su se također razvili prema Jacksonovom tipu. Drugo ime za ovu bolest predložio je profesor N.F. Filatov - “Kozhevnikovska epilepsija”. Četrdesetih godina prošlog vijeka dokazan je odnos EZ s proljetno-ljetnim krpeljnim encefalitisom (ruski encefalitis).
Godine 1958. T. Rasmussen, J. Objevski je opisao kliniku hroničnog fokalnog encefalitisa, čiji je jedan od glavnih simptoma bio EC. Kasnije se bolest zvala Rasmussen encefalitis ili Rasmussenov sindrom (CP). Do sada, ostaje misterija po kojoj bolesti A.Ya. Kozhevnikov je opisao kompleks simptoma EK - u slučaju ruskog encefalitisa ili Rasmussenovog encefalitisa. Po našem mišljenju, A.Ya. Kozhevnikov, koji je praktikovao u Moskvi, opisao je svoj oblik epilepsije, posebno u slučaju hroničnog fokalnog encefalitisa, budući da nijedna od istorija slučajeva koje je on imao nije ukazivao na akutni encefalitis koji su patili od pacijenata.
Pored krpeljnog encefalitisa, EZ uzrokuje tuberkulozni meningoencefalitis, neurosifilis, traumatsku povredu mozga, tumore mozga, fokalnu kortikalnu displaziju, nasljedne metaboličke bolesti.
Hronični fokalni encefalitis [Rasmussenov encefalitis, Rasmussenov sindrom (CP)]. CP predstavlja teške bolesti mozak - hronični progresivni žarišni encefalitis. Bolest se odlikuje trijadom kliničkih kompleksa simptoma: epileptičkim napadima (sličnim kohevnikovom epilepsijom), motoričkim oštećenjima (centralna hemipareza) i poremećajem viših mentalnih funkcija. Etiologija je nepoznata, bolest se verovatno pripisuje sporim neuroinfekcijama virusne etiologije, ali virus nije identifikovan.
Debi u detinjstvu - od 1 godine do 14 godina, sa vrhuncem od 5-6 godina sa epileptičkim napadima (fokalni motor ili sekundarna generalizovana, rjeđe - dijalektička); u 20% slučajeva - sa epilepsijom
tic status. Često obeležena somatosensorna aura (peckanje, peckanje, ukočenost). Već u početnim stadijima bolesti razvija se prolazna postiktalna monopareza (ili hemipareza), Toddova pareza. Obično se nekoliko mjeseci nakon pojave prvih žarišnih napada spajaju dugotrajni (do nekoliko dana), a zatim stalni mioklonični paroksizmi lokalizirani u jednoj polovini trupa i udova, koji se mogu pretvoriti u generalizirane konvulzije. Ovaj kompleksni simptom je Kozhevnikov epilepsija. Vremenom se epileptički mioklonus širi na sve udove, mišiće lica, mišiće prednjeg trbušnog zida i postaje stalan, bez nestajanja čak iu snu. Nastaje perzistentna hemipareza. Prilagođeno kršenje osjetljivosti tipa vodiča i gubitka vidnih polja. Rastuće kognitivno oštećenje, dizartrija. U 25% slučajeva moguća je gojaznost, preuranjeni seksualni razvoj.
EEG u uznapredovalom stadijumu bolesti u 100% slučajeva dolazi do progresivnog usporavanja glavne pozadinske aktivnosti, nastavka regionalnog usporavanja (u frontalno-temporalnim tragovima); nastavak aktivnosti vršnog vala. Kako bolest napreduje, epileptiformna aktivnost se odvija difuzno.
Neuroimaging je presudan u dijagnozi. MRI mozga pokazuje povećanje hemiatrofije u dinamici. Atrofija obično počinje s parijeto-temporalnom regijom u obliku lokalne ekspanzije sylvianskog jaza i širi se vremenom " ulje na listu pergamentnog papira ", hvatajući" zdravu "hemisferu.
EC se odnosi na rezistentne epileptičke sindrome. Početna terapija - valproathe (depakin, concoulex, convulphine) u visokim dozama: do 50-100 mg / kg / dan. Nadalje, preporučuje se kombinacija valproata s levetiracetamom ili topiramatom. Efikasnost levetiracetama sa fokalnim motornim, sekundarno-generaliziranim i miokloničkim napadima u okviru EC, njegova doza je 30-70 mg / kg / dan. Doziranje topiramata je oko 10 mg / kg / dan. U uznapredovalom stadijumu bolesti moguće je koristiti barbiturate (fenobarbital 5-8 mg / kg / dan). Dodavanje etosuksimida (do 30 mg / kg / dan) na početnu AED u nekim slučajevima može biti efikasno u rezistentnim miokloničkim napadima.
Benzodiazepini (klobazam 1 mg / kg / dan ili klonazepam 0,5–4,0 mg / dan) koriste se kod pacijenata sa serijskim napadima i kursom statusa. Propisivanje karbamazepina kao monoterapije se ne preporučuje zbog mogućeg pogoršanja miokloničnih napadaja.
U lečenju samog encefalitisa koriste se različiti lekovi: antivirusni lekovi (zidovudin, aciklovir, ganciklovir); hormonska (metilprednizolon intravenozno 400 mg / m 2 telesne površine 3 dana; prednizon, deksametazon); imunoglobulini (Octagam, IVIC 400 mg / kg / dan intravenski 3 dana); citostatika (azatioprin, ciklofosfamid), plazmafereza. Međutim, ovaj tretman može samo usporiti napredovanje bolesti.
Efektivna neurohirurška intervencija - funkcionalna hemisferotomija, koju treba izvršiti što je ranije moguće. Učestalost stabilne remisije nakon operacije je 23-52%. Bez hirurško liječenje CP napreduje i završava smrtonosno za 2-15 godina (u prosjeku nakon 3 godine) od trenutka debija. Opisani su izolovani slučajevi spontane stabilizacije bolesti.
14.3. Idiopatski generalizovani oblici epilepsije
Benigna mioklonična epilepsija djetinjstva debituje u dobi od 4 mjeseca do 3 godine. Karakteriziraju se isključivo mioklonični napadi u obliku aktivnog mioklonusa u mišićima vrata i proksimalnih dijelova gornjih ekstremiteta: kratki čvorići s blagim naprijed u tijelu, trenutnim podizanjem ramena i laktova u stranu. Obično se javlja serija, koja se povećava nakon buđenja. Svijest nije poremećena. Mnogo rjeđe su primijećeni mioklonični napadaji donji udovi - trenutno savijanje nogu sa blagim čučanjima i čak mogućim iznenadnim padom na zadnjicu.
Mišićna hipotonija i ataksija se otkrivaju u neurološkom statusu. Psihomotorni razvoj ne pati. Na EEG-u se glavna aktivnost ne mijenja; epileptiformna aktivnost se bilježi samo u vrijeme napada. Karakteriziraju ga kratka pražnjenja generalizirane polipo-valne aktivnosti koja se javlja sinkrono sa miokloničkim napadima. Za registraciju kratkih miokloničkih napadaja neophodna je metoda video-EEG monitoringa. Nema promjena u neuroimaging-u.
Starting tretmanizvodi se sa preparatima valproične kiseline. Dodati konvulex ili depakin u sirupu ili kapi (nakon 1-2 godine - preparati tableta) u dozi od 300-1500 mg / dan (15-50 mg / kg / dan). U većini slučajeva dolazi do remisije. U slučaju neefikasnosti, koristi se politerapija; istovremeno valproati ostaju osnovni AEP. Dodelite kombinaciju valproata sa sukcinimidima (etosuksimid u dozi od 250-750 mg / dan, 15-25 mg / kg / dan, u 2-3 doze). Moguće kombinacije valproata sa topiramatom u dozi od 25-100 mg / dan (3-5 mg / kg / dan) u 2 doze; valproat sa benzodiazepinima, na primer, klobazam (frisium) u dozi od 5-20 mg / dan (0,5-1,0 mg / kg / dan) u 2 doze. Propisivanje karbamazepina i lamotrigina je ograničeno zbog mogućnosti pogoršanja miokloničnih napadaja.
Prognozapovoljan. Mentalni razvoj ne pati, a remisija droge se javlja u skoro 100% slučajeva. Trajanje terapije je 3 godine, recidivi su izuzetno rijetki.
Epilepsija sa mioklonija-astatičkim napadima (Dose sindrom) \\ t debituje u rasponu od 1 do 5 godina, češće sa generaliziranim konvulzivnim napadima koji se javljaju u bilo koje doba dana. 11% slučajeva ima povijest febrilnih napadaja. Tipični mioklonični i mioklono-astatički napadaji se obično pridružuju tek nakon 3 godine. Napadi se odlikuju kratkim, munjevitim, obično asinkronim i aritmičnim trzajem u nogama i rukama, često u proksimalnom. Karakteristično je pojavljivanje miokloničnih „čvorova“, kombinovanih sa laganim propulzijama tela i podizanjem ramena („aktivni klimanje glavom“). Učestalost miokloničnih napada može biti vrlo visoka; vrlo često se napadi javljaju opetovano unutar jednog minuta ili čak stalno, naročito nakon buđenja (epileptički status). Kada se mioklonični napadaji javljaju u donjim ekstremitetima, javljaju se kaskadni čučnjevi sa mogućim iznenadnim padom na koljena ili zadnjicu (mioklonični astatski napadi); istovremeno se čuva svest. Abscesi su uočeni u 60-90% bolesnika. Preovlađuju kratki tipični jednostavni apscesi, kao i apsani sa miokloničnom komponentom. Učestalost odsustva je visoka, sa maksimumom ujutro.
U neurološkom statusu su primijećeni jednostrani piramidalni simptomi; u pola slučajeva - grubo
psihorebalno kašnjenje u razvoju. Na EEG-u su otkrivena kratka generalizovana i regionalna pražnjenja aktivnosti vrha i poli-talasa. Promene u snimanju neurona, po pravilu, nisu prisutne; u nekim slučajevima uočena je umerena subatrofija korteksa.
Starting tretmansprovodi se sa preparatima valproinske kiseline u dozi od 600-1750 mg / dan (20-100 mg / kg / dan). Lijek drugog izbora je topiramat u 2 doze u dozama od 50-200 mg / dan (3-7 mg / kg / dan). U slučaju neefikasnosti, koristi se politerapija; međutim, prvo valproati i onda topiramat ostaju osnovni AEP. Koristite kombinaciju valproata sa sukcinimidima, valproatom i topiramatom, valproatom i benzodiazepinima. U nekim rezistentnim slučajevima moguće je dodijeliti tri AED: valproati, topiramat i sukcinimidi (ili benzodiazepini). Upotreba karbamazepina je kontraindikovana zbog mogućnosti pogoršanja miokloničnih napadaja.
Prognoza.Većina dece uspeva da zaustavi napade. Kod otprilike 1/3 pacijenata epileptički napadaji su prisutni, tonički napadi i atipične apsantije, a kognitivni defekt se produbljuje.
Absanse oblici epilepsije. Najčešći i dobro proučavani apsani su deca i adolescenti absansilepsija. Oni se manifestuju kao tipični absani - kratki primarno generalizovani napadaji sa nesvesnim, bledim, minimalnim motoričkim fenomenima i prisustvo na EEG-u simetrične bilateralno sinhrone pik-talasne aktivnosti sa frekvencijom od 3 ili više kompleksa u sekundi (slika 14.2). Postoje jednostavni (blijedi bez motorne komponente) i kompleksni (sa minimalnim motornim fenomenima) izostanci. Komplikacije uključuju izostanke sa tonikom (devijacija leđa, okom gore), mioklonično (trzanje, trzanje kapaka, obrve, nos, ramena), atonsko (padanje glave na grudi, torzo), vegetativno (promjena boje) koža kože, nevoljno mokrenje), kao i asimetrične manifestacije (na primjer, sa blagim zakretanjem glave). Trajanje odsustva napada se kreće od 2 do 30 s, a frekvencija - do 100 ili više dnevno.
Pedijatrijska apscesna epilepsija (pikolepija) - Najčešći oblik odsustva epilepsije. Mutantni geni GABA receptora u
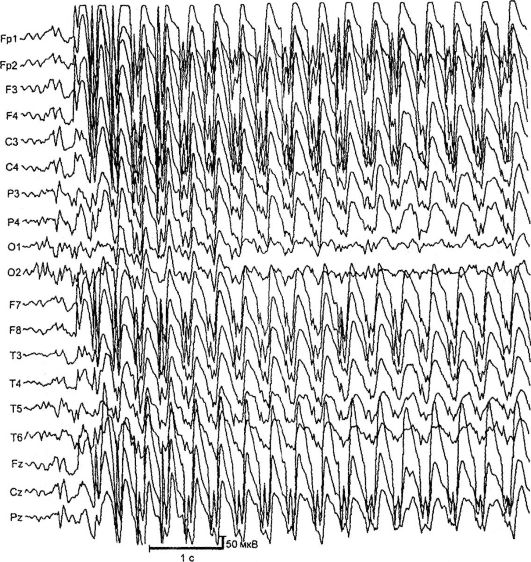
Sl. 14.2.EEG u trenutku napada (odsustvo)
nekoliko lokusa hromozoma: 6p, 8q24, 15q24. Bolest debituje u dobi od 3-9 godina sa tipičnim izostancima. U retkim slučajevima, bolest počinje generaliziranim konvulzivnim napadima, nakon čega slijedi dodavanje izostanaka. Češće su bolesne djevojke. Tipičan tip napadaja je absans sa toničnom komponentom: blagim padanjem glave i uspostavljanjem očnih jabučica. Napadi su izazvani hiperventilacijom, rjeđe - oralnim brojanjem. Uz neadekvatan tretman, oko 30% pacijenata se pridružuje SHG-ovima. Neprekidna generalizovana pražnjenja sa frekvencijom od 3 Hz pojavljuju se na EEG-u tokom hiperventilacije. MRI promjene se ne otkrivaju.
Anabolička aktivnost ima: valproat, sukcinimid, benzodiazepin, lamotrigin, topiramat. Upotreba droge
karbamazepin je kontraindikovan jer izaziva povećanje napadaja. Početni tretman se vrši preparatima valproinske kiseline 2 puta dnevno, u dozi od 600-1800 mg / dan (30-50 mg / kg / dan). Kod većine pacijenata, napadi potpuno prestaju sa monoterapijom valproatom. Drugi izbor lijekova - sukcinimidi. Sukcinimidi se koriste kao monoterapija kada pacijent ima izolovane izostanke, doza etosuksimida je 500-1000 mg / dan (15-30 mg / kg / dan) u 3 doze.
U rijetkim rezistentnim slučajevima koristi se politerapija: valproati + sukcinimidi, valproati i lamotrigin. Potpuna terapijska remisija se postiže u 90-97% slučajeva, obično uz monoterapiju. Povlačenje lekova počinje 3 godine nakon prestanka napada.
Epilepsija juvenilnog apscesa (UAE) - oblik idiopatskog generalizovanog oblika epilepsije, karakteriziran tipičnom odsutnošću, debutirajući u pubertetskom periodu s velikom vjerovatnoćom spajanja GSP i EEG promjene u obliku kratkih pražnjenja generalizirane brze aktivnosti vrha. Etiologija je mutacija gena nikotinskog acetilkolinskog receptora povezanog s kromosomima 5, 8, 18 i 21. Bolest počinje u dobi od 9-21 godina (maksimalno - tijekom puberteta). U 40% slučajeva epilepsija debituje sa SHG, u ostalom - sa odsustvom. Karakterišu ga jednostavni apsani, kraće trajanje i učestalost nego u dečjem obliku. U nekim slučajevima se nalaze vrlo kratke (do 3 s) izostanci sa miokloničnom komponentom: blijeđenje, lako postavljanje očne jabučice prema gore i brzo trzanje kapaka. Kod 75% pacijenata je uočena kombinacija odsutnosti sa SHGs. Konvulzivni napadaji se obično javljaju ujutro nakon što se pacijenti probude. Učestalost napada je mala - 1-4 puta godišnje.
EEG se odlikuje normalnom primarnom aktivnošću, na čijoj se osnovi detektiraju kratka pražnjenja generalizirane brze (4 Hz) vršne aktivnosti. Pojava epileptiformne aktivnosti tokom deprivacije sna, ritmička fotostimulacija i zatvaranje očiju je od velike dijagnostičke važnosti. Sa JAE fotosenzitivnost je 20,5%, a kod AED - 10%. Test sa hiperventilacijom u slučaju UAE je neinformativan.
Početna terapija se vrši lekovima valproične kiseline u dozi od 900-2000 mg / dan (30-40 mg / kg / dan) u 2 doze. Sa
u odsustvu efekta monoterapije, prelaze na kombinovanu terapiju (valproat + topiramat, valproat + sukcinimid).
Potpuna terapijska remisija se postiže u prosjeku kod 70% pacijenata. Terapija otkazivanja se sprovodi postepeno, ne manje od 4 godine od potpunog odsustva napada.
Epilepsija sa izolovanim generaliziranim konvulzivnim napadima (epilepsija sa generaliziranim konvulzivnim napadima buđenja) (EGSP) je oblik idiopatske generalizovane epilepsije, u kojoj su jedini tipovi napada primarno generalizirani toničko-klonični konvulzivni paroksizmi bez aure i jasan fokus na EEG. Forma je određena genima CLCN2 na hromozomu 3q26 i genomu CACNB4 na kromosomu 2q22-23.
Debut bolesti u širokom rasponu godina - od 10 do 30 godina (maksimalno - u pubertetskom periodu). Generalizovani toničko-klonični napadi javljaju se bez aure, ograničeni na period buđenja ili zaspanja. To je izazvano deprivacijom sna (smanjenje ukupnog trajanja sna, kasnog spavanja, buđenja u neuobičajeno ranom vremenu). Trajanje SHG-ova je od 30 s do 10 min, njihova frekvencija je mala. Većina pacijenata nema više od 2-5 napadaja godišnje.
EEG u interiktalnom periodu kod 50% pacijenata je normalan. Preporučuju se EEG nakon deprivacije sna i noćnog video EEG monitoringa. U interiktalnom periodu dolazi do kratkog generalizovanog pražnjenja. Toničnu fazu SHG karakteriše pojava difuznog EEG-a, a amplituda se povećava frekvencijom brzog ritma od 20-40 Hz, postepeno usporavajući na 10 Hz. Tokom klonske faze, ovaj ritam se postepeno zamenjuje generalizovanom polip-talasnom aktivnošću. U fazi opuštanja nakon napada, dominantna je difuzna delta aktivnost; regionalni fenomeni su odsutni.
U EGSP-u postoji visoka efikasnost svih glavnih AEP grupa: barbiturati, hidantoini, karbamazepin, okskarbazepin, valproati, topiramat, levetiracetam. Fenobarbital i difenin zbog izraženog nuspojave primjenjuju se na posljednjem mjestu u odsustvu efekta osnovnog AEP-a. Osnovni lekovi za epilepsiju sa GSP su topiramat, valproati i grupa karbamazepina.
Liječenje počinje topiramatom u dozi od 100-400 mg / dan (4-10 mg / kg / dan) u 2 podijeljene doze. Drugi lijek je valproična kiselina u dozi od 1000-2000 mg / dan (30-50 mg / kg / dan) u 2 doze. Lijek trećeg izbora je karbamazepin ili okskarbazepin (trileptal).
U nekim rezistentnim slučajevima moguća je monoterapija barbituratima ili hidantoinima, koja efikasno, ali često dovodi do razvoja izraženih nuspojava i smanjuje kvalitet života pacijenata. U rijetkim rezistentnim slučajevima, potrebno je pribjeći polieterapiji. Optimalna kombinacija: topiramat + valproati; u isto vrijeme doze lijekova ostaju nepromijenjene.
Remisija se postiže kod 90% pacijenata. Nedostatak efekta često je povezan sa pogrešnom dijagnozom. Uz neadekvatan tretman, moguće je pristupanje izostanaka ili mioklonusa sa transformacijom u SAE i UME.
Juvenilna mioklonična epilepsija (UME - Janzov sindrom) je oblik idiopatske generalizovane epilepsije, karakteriziran debutom u adolescenciji i prisustvom masivnih miokloničnih napadaja koji se javljaju pretežno u periodu nakon buđenja pacijenata.
YuME je heterogena bolest povezana sa mutacijom nekoliko gena, uključujući i GABRA1 gen(OMIM 137160) na kromosomu 5q34-q35, CACNB4 gen(OMIM 601949) na kromosomu 2q22-q23 i mutaciji CLCN2gen (OMIM 600570) na kromosomu 3q26. Rizik od epilepsije kod dece u porodici gde jedan od roditelja ima UM je oko 8%. Generalizirana aktivnost vršnih valova na EEG nalazi se u 18% klinički zdravih rođaka probanda koji pate od UME.
Bolest počinje u dobi od 7 do 21 godine s maksimumom u dobnom rasponu od 11-15 godina. Glavni tip napadaja su mioklonični paroksizmi, koji se odlikuju brzim trzanjem različitih mišićnih grupa. Oni su češće bilateralni, simetrični, pojedinačni ili višestruki, varirajući u amplitudi; često se pojavljuju kao serija volja. Lokaliziraju se uglavnom u ramenom pojasu i rukama, uglavnom u grupama savijenih mišića. Spašena je svijest tijekom miokloničkih napada. Kod 30% pacijenata mioklonični napadaji zahvaćaju mišiće nogu, dok pacijent osjeća iznenadan udarac ispod koljena i blago čučnjeva ili padova (mioklonični astatski napadi). Do miokloničnih napadaja dolazi ili
povećanje u prvim minutama i satima nakon buđenja. Smanjena budnost, pospanost, zijevanje, prekrivanje očiju povećava vjerovatnoću napada ujutro.
U 90% slučajeva mioklonični napadaji se kombinuju sa GSP buđenjem - ovaj tip napada naziva se kloničko-toničko-klonski. U 40% bolesnika pridružuju se kratke odsutnosti.
Provokativni faktori su deprivacija sna i naglo nasilno buđenje. Kod nekih pacijenata mioklonični napadaji se javljaju samo kada nema dovoljno sna. Približno 1/3 pacijenata sa UME (obično ženskim) napadima su fotosenzitivni: izazvani gledanjem televizije, kompjuterskih igara, treperećih svjetala u diskotekama. Glavni EEG uzorak su kratki ispadi generalizovane brze polipične valne aktivnosti, otkriveni u 80-95% bolesnika u interiktalnom periodu. Najtipičnija je generalizovana brza (4 Hz i veća) polip-valna aktivnost. EEG sa UME treba obaviti rano ujutro nakon noći deprivacije sna.
Diferencijalna dijagnoza UME se izvodi sa tikovima, korejom, kao i sa različitim oblicima progresivne epilepsije sa mioklonusom. Uz terapiju lijekovima, strogo se mora poštivati režim sna i budnosti; izbjegavajte faktore stimulacije fotomodeliranja u svakodnevnom životu.
Početni tretman - lekovi valproične kiseline u dozi od 1000-2500 mg / dan (30-50 mg / kg / dan). Kako bi se izbjegle nuspojave kod djevojaka (menstrualni poremećaji, gojaznost, hirzutizam, policistični jajnici, smanjena plodnost), liječenje se može započeti s topiramatom ili levetiracetamom kao monoterapijom. Topiramat se propisuje u dozi od 200-400 mg / dan (5-10 mg / kg / dan) u 2 doze. Levetiracetam se daje u dozi od 30-60 mg / kg / dan
(1000-3000 mg / dan) u 2 doze.
U slučaju nedovoljne efikasnosti propisuje se polieterapija: valproati + sukcinimidi (sa rezistentnim apsanima); valproat + topiramat ili levetiracetam (za rezistentne SHG); valproat + benzodiazepini (sa izraženom fotosenzitivnošću). Preparati karbamazepina su kontraindikovani.
Kompletna remisija leka se postiže kod 85-95% pacijenata, au većini slučajeva primjenom monoterapije. Problem je visoka stopa recidiva nakon ukidanja AED-a. Ukidanje lekova, čak i nakon 4-5 godina potpune kliničke remisije, uzrokuje
ponavljanje napadaja kod najmanje 50% pacijenata. Preporučuje se da AEP bude ukinut najranije nakon 4 godine bez napada.
14.4. Epileptička encefalopatija djetinjstva i djetinjstva
Westov sindrom - simptomatski ili kriptogeni oblik generalizirane epilepsije, karakteriziran djelima infantilnih spazama, hiparitmijom na EEG, odgođenim psihomotornim razvojem. Bolest debituje u prvoj godini života, uglavnom u dobi od 6-8 mjeseci. Glavni tip napadaja su infantilni fleksorni grčevi ("Salaamovi napadi"): dete savija glavu i torzo, podiže i savija ruke i noge. Napadi su veoma kratki, sekundi; često grupisane u seriju - do 100 ili više grčeva po 1 seriji. Do 10–50 epizoda sa češćim pojavama nakon buđenja prati se dnevno. U nekim slučajevima, moguća je izražena asimetrija grčeva, u drugima proširenje trupa i ekstremiteta (ekstenzorski tonički spazam). Često se uočava izraženo odlaganje psihomotornog razvoja i tetrapareze. U simptomatskim slučajevima, promjene u neurološkom statusu se otkrivaju ubrzo nakon rođenja; sa kriptogenim - samo sa početkom napada.
EEG se odlikuje difuznom neredovnom visokomakularnom sporom talasnom aktivnošću sa slabo primetnom spike komponentom, hipsaritmijom. Moguća asimetrija epileptiformnih obrazaca i njihova dominacija u okcipitalnim potezima (Sl. 14.3).
Neuroimaging se karakteriše difuznom atrofijom, malformacijama mozga, efektima perinatalne encefalopatije. Kao poseban uzrok razvoja bolesti izolovana je tuberkularna skleroza, kao i neka nasledna degenerativna i metabolička oboljenja.
Neophodna je rana primjena lijeka za infantilne grčeve. Početna terapija počinje vigabatrinom (sabrila) - 50-100 mg / kg / dan ili valproat - 50-100 mg / kg / dan. Topiramat (topamax) u dozi od 5-10 mg / kg / dan može biti lijek drugog ili trećeg izbora. Za rezistentne konvulzije, kombinacija ovih osnovnih AED-a sa benzodiazepinima (klonazepam 0,25–2 mg / dan, klobazam 1 mg / kg / dan) ili fenobarbitalom (5–15 mg / kg / dan), kao i sa suksilepom (15-30 mg / kg / dan). U asimetričnim napadima može se dodati karbamazepin (finlepsin, tegretol) u dozi od 10-20 mg / kg / dan.
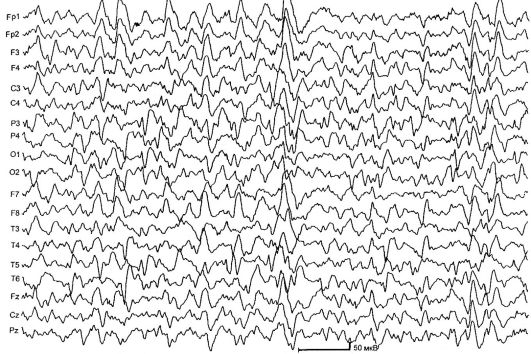
Sl. 14.3.EEG sa Westovim sindromom (gipsaritmija)
Alternativni metod je upotreba kortikosteroidnih hormona (sinakten-depot intramuskularno; deksametazon, prednizolon oralno) i imunoglobulina (oktagam). Prosečna doza prednizona je 1-2,5 mg / kg / dan, a zatim prelazi na minimalnu dozu održavanja. Hormoni se obično propisuju u kombinaciji sa osnovnom AED. Lečenje steroidima obavljaju specijalisti u klinici zbog opasnosti od ozbiljnih neželjenih efekata.
Prognoza je teška. Moderni AED omogućavaju zaustavljanje napadaja kod 60% pacijenata, ali u većini slučajeva ostaje izražen intelektualni defekt i autistično slično ponašanje. Sa perzistencijom napadaja, primećena je transformacija u tešku multifokalnu epilepsiju ili Lennox-Gastautov sindrom.
Lennox-Gasto sindrom (dječja epileptička encefalopatija s difuznim usporenim valovima na EEG-u) (SLH) - kriptogena (simptomatska) generalizirana epilepsija, koju karakterišu česti polimorfni napadi, specifične promjene u EEG-u, smanjena inteligencija, otpornost na terapiju. Etiologija je u većini slučajeva nepoznata. SLH je jedan od najtežih oblika epilepsije.
Bolest se debitira češće u dobi od 3 do 8 godina. Karakterizira ga trijada napada, koja se uočava u gotovo 100% slučajeva: tonički aksijalni, atipični izostanci i napadi padova. Tonički napadaji se manifestuju kratkom intenzivnom napetošću muskulature trupa i ekstremiteta, koji se češće javljaju noću. Ponekad su duži, praćeni laganim kloničnim trzanjem udova (tonički-vibracijski napadi) i teškim autonomnim simptomima (apneja, bradikardija). Atipična apsancija karakteriše postepeniji početak i kraj napadaja nego kod tipičnih absana; svijest često fluktuira; tu su atonični fenomeni (pad glave na grudi, spuštanje ramena, savijanje tela, rezanje nogu). Napadi padova mogu imati oštar tonički karakter ("pasti statuom") ili glađi - mijatonski (početna mioklonična komponenta, zatim atonija). Za vrijeme ovih padova, djeca trpe razne ozljede glave i torza. U nekim slučajevima, uočeni su mioklonični i generalizirani konvulzivni napadaji; pojava fokalnih napada je predmet diskusije. Karakterizira ga najveća učestalost napada sa povećanjem sna, na buđenju, u periodu pasivne budnosti. Naprotiv, aktivna budnost doprinosi smanjenju napada ["moždana aktivnost je antagonizam napadaja" (Gastaut)]. Kod bolesnika sa SLH postoji velika vjerovatnoća pojave serijskih napada i epileptičkog statusa (tonički napadaji i abnormalni apsani). Status toničkih napada može predstavljati neposrednu opasnost za živote pacijenata.
U neurološkom statusu utvrđena je difuzna hipotenzija, ataksija. Simptomi poraza piramidalnih načina, po pravilu, nisu prisutni. Intelekt je smanjen u svim slučajevima; može doći do hiperaktivnog, autističnog ili psihopatskog ponašanja.
Na EEG-u su identifikovana 3 glavna obrasca: usporavanje glavne aktivnosti pozadinskog snimanja, sporo difuzni kompleksi, akutno-spor val, brza (10-20 Hz) aktivnost, najčešće tokom spavanja (slika 14.4).
Neuroimaging ne otkriva lokalne strukturne defekte mozga; u većini slučajeva utvrđuje se difuzna kortikalna atrofija.
Tretman je prikazan u tabeli. 23.
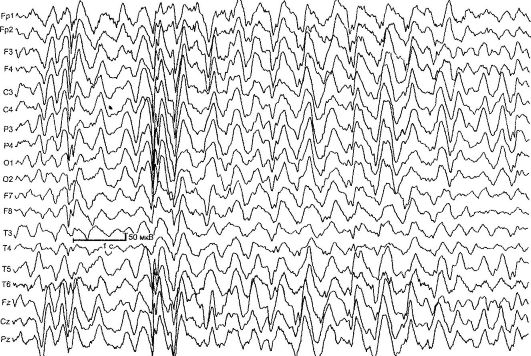
Sl. 14.4.EEG sa Lennox-Gasto sindromom
Tabela 23.Tretiranje Lennox-Gasto sindroma
Anti-epileptički lekovi zauzimaju vodeće mesto u lečenju SLH; sve ostale metode su pomoćne. Počinje terapija topiramatom. Njegova početna doza je obično
12,5 mg / dan. Da bi se izbjegle moguće nuspojave, pokazana je usporena titracija doze - povećanje od 12,5 mg svake sedmice. Doziranje topiramata je 75-350 mg / dan (3-10 mg / kg / dan) i veće u 2 doze. Drugi izbor lijeka - valproična kiselina. Preparati valprojske kiseline propisuju se postepenim povećanjem do 900-2500 mg / dan (40-80) mg / kg / dan i više do maksimalno tolerisane doze.
Uz nedovoljan efekat monoterapije (u većini slučajeva) preporučuje se prelazak na kombinaciju lekova: topiramat + valproat, valproat + sukcinimid, valproat ili topiramat + lamotrigin. Sukcinimidi se koriste u dozi od 500-1000 mg / dan (20-35 mg / kg / dan) u 3 doze. Lamotrigin počinje sa 12,5 mg / dan, povećavajući dozu od 12,5 mg jednom nedeljno; prosječna doza lijeka je 75-200 mg / dan (3–7 mg / kg / dan) u 2 doze.
Sa tonički epizodama koje su otporne na tretman i na bazne AED, može se dodati karbamazepin. U ovim slučajevima, optimalna šema je valproat + karbamazepin. Karbamazepine treba propisati u malim ili srednjim dozama i samo u kombinaciji sa osnovnim AEP. Prosečna doza karbamazepina je 100-600 mg / dan (10-20 mg / kg / dan) u 2 doze. Levetiracetam u dozi od 1000-3000 mg / dan (30-60 mg / kg / dan) može biti efikasan u miokloničkim i generaliziranim konvulzivnim napadima. Sa učestalošću toničkih napada, moguća je kombinacija valproata i hidanotoina. Difenin se primjenjuje u dozi od 75-200 mg / dan (3-7 mg / kg / dan) u 2 doze.
U odsustvu efekta terapije u terapijskom režimu, moguće je uvesti benzodiazepine u kombinaciji sa osnovnim AEP. Od benzodiazepina, samo se klobazam može koristiti za dugoročno liječenje pacijenata sa SLH. Clobazam se daje u dozi od 10-30 mg / dan (0,5-1,0 mg / kg / dan). Svi ostali benzodiazepini treba da se daju oralno samo kao "lekovi protiv požara" za nekontrolisano serijsko povećanje napadaja.
Najčešća kombinacija u rezistentnim napadima kod pacijenata sa SLH je topiramat + valproat + sukcinimid (ili klobazam).
Prognozasa SLH nepovoljnim. Samo 5-15% pacijenata uspijeva postići remisiju. U drugim slučajevima, terapija modernim AEP-om omogućava da se smanji učestalost napada, da se izbjegne pojava epileptičkog statusa i smanji intelektualna i mnemonička
nedostatak. Očekivano trajanje života zavisi od brige o pacijentu. Većina pacijenata je duboko onesposobljena, nesposobna za samostalan život.
Landau-Kleffner-ov sindrom [stečena epileptička afazija (SLK)] - pretpostavlja se idiopatski oblik epilepsije. Po prvi put, elektroklinička slika bolesti opisali su V. Landau i F. Klefner 1957. godine. Ovo je prilično rijedak oblik dječje epilepsije, koji se manifestira stečenom senzomotornom afazijom u kombinaciji s različitim epileptičkim napadima i difuznim promjenama u EEG-u. SLK se pojavljuje u dobi od 3-7 godina. Do početka bolesti motorički, mentalni i govorni razvoj pacijenata je prikladan uzrast.
Poremećaji govora - kardinalni znak bolesti. Često se razvijaju postepeno, nekoliko sedmica ili mjeseci, rjeđe - katastrofalno brzo, u roku od nekoliko dana. Prvi simptom bolesti obično je istog tipa: roditelji primjećuju da dijete adekvatno prestaje reagirati na adresirani govor (manifestacije senzorne afazije). Tokom ovog perioda mogu se javiti naglašeni poremećaji ponašanja: emocionalna labilnost, razdražljivost, hiperaktivnost; izraženi negativizam, epidemije agresije. Pojavljuju se daljnje povrede ekspresivnog govora: pacijenti počinju govoriti jednostavnim izrazima, onda koriste samo odvojene riječi i prestaju da govore.
Drugi simptomski kompleks SLK - epileptički napadaji. Karakteristični fokalni motorički napadi (faringeoralni i hemifacijalni), kao i atipične izostanke. Manje su uobičajeni atonični, mioklonični i generalizirani konvulzivni paroksizmi. U većini slučajeva napadaji su rijetki; opaženo prilikom zaspanja i buđenja. U 1/4 bolesnika nema epileptičkih napadaja. U tim slučajevima dijagnoza se utvrđuje na osnovu pojave stečene afazije, izraženog kognitivnog oštećenja i EEG podataka.
Fokalni simptomi ne postoje u neurološkom statusu. Psihološko testiranje otkriva senzornu ili totalnu afaziju, povrede prakse. Karakteristični poremećaji ponašanja.
EEG određuje prisustvo epileptiformnih poremećaja u 100% slučajeva. Tipični su regionalni oštri talasi visoke amplitude (200-400 µV) ili kompleksi akutnog sporog talasa, lokalizovani
kupaonice uglavnom u temporalnim ili parijetalno-temporalnim predjelima. Epileptiformna aktivnost se povećava u snu (u fazi i brzog i sporog sna), širi se difuzno, obično održavajući amplitudnu dominaciju dominantne hemisfere za govor. U određenim epohama snimanja u snu, indeks epileptiformne aktivnosti može doseći 100%. To je epileptiformna aktivnost koja dovodi do razvoja teških govornih poremećaja (manifestacija kognitivne epileptiformne dezintegracije). MRI je obično normalan.
Režim liječenja za SLK ovisi o prisutnosti ili odsutnosti epileptičkih napadaja. Kada je SLK bez epileptičnih napadaja, monoterapija sa sukcinimidima ili benzodiazepinima je efikasna. Početni tretman se izvodi sa sukcinimidima. Etosuksimid se propisuje u dozi od 500-1000 mg / dan (25-35 mg / kg / dan) u 3 doze. Lijek drugog izbora - klobazam u dozi od 10-30 mg / dan (0,5-1,0 mg / kg / dan) u 2-3 doze. Ovi lekovi blokiraju kontinuiranu difuznu epileptiformnu aktivnost na EEG, što dovodi do oporavka govornih funkcija. U prisustvu epileptičkih napadaja, koriste se samo kao dodatni AED.
Kod SLK sa epileptičkim napadima, tretman počinje valproičnom kiselinom u dozi od 900-2000 mg / dan (30-70 mg / kg / dan) u 2 doze. Lijek drugog izbora je topiramat. Topamax se primenjuje uz postepeno povećanje doze do 50-150 mg / dan (3-7 mg / kg / dan) u 2 doze. Uz neefikasnost monoterapije treba nastaviti s kombiniranim liječenjem. Optimalne kombinacije sa SLK: valproati + sukcinimidi, valproati + topiramat, valproati + benzodiazepini. Jedan od najvažnijih kriterijuma za efikasnost terapije je blokiranje fenomena sekundarne bilateralne sinhronizacije na EEG (difuzna pražnjenja).
Upotreba karbamazepina je kontraindicirana s obzirom na mogući porast napadaja, poboljšanje sekundarne bilateralne sinhronizacije na EEG i produbljivanje govornih poremećaja.
Kortikosteroidi (synacthen depot, deksametazon) su rezervni lijekovi. Oni pozitivno utiču na oporavak govora. Moguća pulsna terapija deksametazonom u dozi od 1 mg / kg / dan. Metoda se sastoji u imenovanju lijeka svaka 2 tjedna; onda je interval bez deksametazona 4-8 nedelja, pa opet 2 nedelje. U ovom slučaju, osnovna terapija AED-a se izvodi bez prekida.
U kvalitetu hirurško liječenje sa SLK-om primijeniti podbradne zareze.
Prognozau slučaju SLK postoje povoljni epileptički napadi: u 100% bolesnika, napadi se potpuno zaustavljaju u pubertetu (pod uticajem AEP ili spontano). Međutim, u odsustvu terapije ili neadekvatnog liječenja (možda s neprepoznatom epileptičnom prirodom bolesti), govorno i kognitivno oštećenje može postojati.
Epilepsija sa električnim epileptičkim stanjem sna (sinonimi: epilepsija sa kontinuiranom aktivnošću vršnog vala na EEG-u tokom sporog sna, ESES-sindrom - električni status epilepticus tokom spavanja) prema klasifikaciji iz 1989. odnosi se na oblike koji imaju karakteristike i generaliziranog i parcijalnog. Patogeneza sindroma povezana je sa stalnim “bombardovanjem” kontinuiranom epileptiformnom aktivnošću kortikalnih centara sa razvojem njihove funkcionalne inhibicije i rupture neuronskih veza, što dovodi do razvoja teškog kognitivnog oštećenja.
Patognomonsko prisustvo fokalnih i pseudo-generalizovanih epileptičkih napadaja u kombinaciji sa teškim kognitivnim oštećenjem i obrascem kontinuirane difuzne epileptiformne aktivnosti tokom perioda sporog sna, nastavljajući kontinuirano mnogo mjeseci i godina.
Postoje idiopatske i simptomatske varijante sindroma. U simptomatskoj varijanti, odloženom psihomotornom razvoju, fokalnim neurološkim simptomima (strabizam, hemiparetična cerebralna paraliza, ataksija), strukturne promjene u neuroimagingu prisutne su prije početka napada. U “klasičnoj” (idiopatskoj) verziji ovi znakovi su odsutni. Debitno doba epileptičnih napadaja varira, prema Tassinari (2002), od 8 mjeseci do 12 godina, u prosjeku 4,7 godina. Među pacijentima sa dječacima prevladavaju. Ne manje od 1/3 pacijenata ima epileptičke napade. U ovom slučaju, dijagnoza se uspostavlja na osnovu kombinacije kontinuirane, kontinuirane epileptiformne aktivnosti u sporom spavanju sa teškim kognitivnim oštećenjem.
Početak bolesti s fokalnim motoričkim (faringeoralnim, hemifacijalnim, unilateralnim) napadima ili naizmjeničnim hemikonvulzijama, koje se javljaju pretežno
vrijeme spavanja (posebno prije buđenja). U 15% slučajeva utvrđena je anamneza febrilnih napadaja. Napadi su obično rijetki; u nekim slučajevima - pojedinačno. U ovoj fazi nema izraženog kognitivnog oštećenja. Tokom ovog perioda bolesti, dijagnoza se ne može utvrditi.
Drugi period (razvijene kliničke manifestacije) dolazi za nekoliko meseci ili godina od trenutka debi prvog napada. Klinički je karakteriziran pojavom „pseudo-generaliziranih“ napadaja i, prije svega, atipičnih izostanaka, obično s atonskom komponentom („klimanje“, savijanje naprijed, savijanje nogu). Osim toga, mogući su i mioklonični napadaji, paroksizmalni padovi i generalizirani toničko-klonički napadaji. Većina ovih napada - rezultat fenomena sekundarne bilateralne sinhronizacije na EEG-u. S pojavom ovog fenomena, kognitivna oštećenja postaju vidljiva i ubrzano se povećavaju. Poremećaji kognitivnih funkcija (pamćenje, pažnja, brzina reakcije, izvršenje komandi, itd.) S oslabljenom socijalnom adaptacijom i nemogućnošću učenja nazivaju se „dječja epileptiformna kognitivna dezintegracija“. Promjene u ponašanju (psihopatski, shizofreni, autistični sindromi). Poremećaji govora uključuju senzornu ili motoričku afaziju, orolingvomukomotornu dispraksiju, slušnu agnoziju. Postoji perzistentna hemipareza ili ataksija (kada se epileptički fokus nalazi uglavnom u motornom korteksu). Rijetki simptomi uključuju alexia, acalculus. U većini slučajeva, sve vrste kršenja su kombinovane u jednom ili drugom stepenu. Pojava u klinici bolesti „pseudo-generaliziranih“ napadaja i oštećenja viših mentalnih funkcija korelira s pojavom EEG-a u nastavku epileptiformne aktivnosti u sporom spavanju.
U trećoj, završnoj fazi, učestalost napada se postepeno smanjuje; postaju retki, izolovani, osetljiviji na terapiju. U isto vrijeme postoji postepeno stabilno poboljšanje viših mentalnih i motoričkih funkcija (obično s početkom puberteta).
EEG igra ključnu ulogu u dijagnostici EESM-a. Možda odsustvo epileptiformne aktivnosti tokom budnosti. Karakterizira se pojavom i naglim povećanjem difuzne epileptiformne aktivnosti u periodu sporog spavanja s najvišim indeksom, koji u ovoj fazi doseže 85-100%. Ova aktivnost
traje stalno mnogo mjeseci i godina. Fiziološki obrasci sna nestaju. Tokom REM spavanja, epileptiformna aktivnost je smanjena ili blokirana.
Neuroimaging metode u većini slučajeva ne otkrivaju nikakve abnormalnosti. U simptomatskim slučajevima primijećena su lokalna oštećenja nastala kao posljedica perinatalnog oštećenja i disgeneze mozga.
Taktički tretman zavisi od prisustva ili odsustva epileptičkih napadaja u sindromu EESM. U električnom epileptičkom statusu sporog spavanja bez epileptičkih napada, efikasna je monoterapija sa sukcinimidima ili benzodiazepinima. Etosuksimid se propisuje u dozi od 500-1000 mg / dan (25-35 mg / kg / dan) u 3 doze. Lijek drugog izbora su benzodiazepini. Klobazam se koristi u dozi od 10-30 mg / dan (0,5-1,0 mg / kg / dan) u 2-3 doze. Ovi lekovi oštro blokiraju kontinuiranu difuznu epileptiformnu aktivnost na EEG i indirektno dovode do poboljšanja kognitivnih funkcija.
U prisustvu epileptičkih napadaja, koriste se samo kao dodatni AED, a započinjanje terapije se vrši preparatima valproične kiseline, a zatim i monoterapijom topiramatom. Valproati se propisuju u dozi od 600-2000 mg / dan (30-70 mg / kg / dan) u 2 doze. Lijek drugog izbora, topiramat, primjenjuje se postepenim povećanjem doze do 50-150 mg / dan (3-7 mg / kg / dan) u 2 doze.
U slučaju nedovoljne efikasnosti monoterapije primenjuje se kombinovani tretman. Optimalne kombinacije: valproati + sukcinimidi, valproati + topiramat, valproati + benzodiazepini (klobazam). Najvažniji kriterijum za efikasnost lečenja je smanjenje indeksa ili potpuno blokiranje nastavka aktivnosti epileptifora na EEG. Upotreba karbamazepina je kontraindikovana zbog mogućnosti pojave ili povećanja napadaja, kao i poboljšanja sekundarne bilateralne sinhronizacije na EEG i produbljivanja kognitivnog oštećenja.
U rezistentnim slučajevima, kortikosteroidi (synacthen depot, prednizon, metipred, deksametazon, itd.) Moraju biti dodani osnovnom AEP. Sinakten depot se propisuje počevši od 0,1 mg / dan, postepeno povećavajući se na 0,1 mg svakih 3-5 dana na 1,0 mg / dan. Trajanje liječenja je od 3-4 tjedna do nekoliko mjeseci uz postupno otkazivanje. U ovom slučaju, osnovna terapija AEP
održan bez prekida. Hormoni imaju izražen blokirni efekat na aktivnost epileptifora na EEG i doprinose poboljšanju govornih funkcija.
U slučaju simptomatske prirode, moguće je hirurško liječenje - kortikalna resekcija. Na primer, sa hemimegall cefalijom, jedini način da se izbegne teška ireverzibilna kognitivna dezintegracija je funkcionalna hemisfera.
Prognoza je pogodna za epileptičke napadaje i ozbiljna za kognitivna oštećenja. Napadi dobro reaguju na adekvatnu AED terapiju i obično nestaju nakon 10-12 godina. Uz postepeni nestanak kontinuirane epileptiformne aktivnosti u fazi spavanja, kognitivne funkcije se također poboljšavaju do početka puberteta. Međutim, polovina pacijenata „izlazi“ iz bolesti sa izraženim intelektualno-mentalnim defektom i nesposobni su da uče u srednjoj školi.
Specifični sindromi. Među specifičnim sindromima u pedijatrijskoj neuroznanosti, posebno su relevantni febrilni napadaji i status epilepticus.
14.5. Febrilni napadi
Febrilni napadi (FS) - konvulzije kod djece od 6 mjeseci do 5 godina, koje se javljaju na temperaturi koja nije povezana s neuroinfekcijom. Konvulzije kod djece u akutnom stadiju meningitisa, encefalitisa nisu klasificirane kao FS, ali se smatraju simptomatskim manifestacijama neuroinfekcije. FS se javlja kod 5% djece. Osnova FS je genetski determinisano smanjenje praga konvulzivne spremnosti sa pojavom generalizovanih konvulzivnih pražnjenja tokom hipertermije. Poligensko nasleđivanje; pretpostavljeni su defekti u FEB1 lokusu (hromozom 8ql3-q21) i FEB4 na 5ql4ql5 kromosoma. Vjerovatnoća pojave FS kod djece, ako je jedan od roditelja imala istoriju, može biti prilično visoka - 5-20%.
Dodjeljivanje tipičnog (jednostavnog) i atipičnog (kompleksnog) FS. Tipični FS su genetski određeni, javljaju se u neurološki zdravoj djeci i čine do 90% svih slučajeva FS. Oni se manifestuju generalizovanim toničko-kloničkim napadima na pozadini visoke temperature. FS se obično javlja prvog dana povišenja temperature, obično kada dijete zaspi. Trajanje
napadi ne prelaze 10 minuta, simptomi gubitka nakon napada su odsutni. EEG u interiktalnom periodu unutar normalnih granica. Više od 50% djece ima FS. Tipični FS ne utiče na razvoj djeteta i prolazi bez liječenja nakon 5 godina bez traga. Rizik transformacije u epilepsiju (uglavnom idiopatske forme) ne prelazi 10%.
Atipični FS (kompleksni) čine oko 10% svih slučajeva FS. Karakterišu ih sledeće karakteristike.
Debut starosti do 1 godine ili nakon 5 godina.
Veliko trajanje napada - više od 30 minuta.
Prvi napad može pokazati epileptički status, koji zahtijeva reanimaciju.
Napadi sa fokalnom komponentom: adverzija glave i očiju, hemikonvulzije, “šepanje”.
Početak simptoma gubitka nakon napada (na primjer, Toddina paraliza, afazija).
Identifikovani su fokalni neurološki simptomi i kognitivna oštećenja.
Regionalno usporavanje EEG-a u jednom od vremenskih vodova.
MRI identificira značajan simptom atipičnog FS - mesijalne temporalne skleroze (rezultat ishemijskog moždanog udara s dugotrajnim FS, prekrivenim nezrelim mozgom).
Atipični FS imaju lošu prognozu. Često su u kombinaciji sa kognitivnim oštećenjem. Rizik transformacije u epilepsiju je oko 15%; u prisustvu mesijalne temporalne skleroze - dramatično se povećava. Dugotrajna netipična atipična FS može dovesti do razvoja akutnog oštećenja cerebralne cirkulacije ishemijskog tipa i pojave perzistentnog motornog deficita. U ovom slučaju, nakon epizode FS, razvija se hemipareza i rezistentna epilepsija sa hemikonvulzivnim napadima: HHE sindrom (epilepsija sa hemikonvulzijama i hemiplegijom).
Pacijenti sa tipičnim FS ne trebaju dugotrajnu terapiju i prevenciju droga. Preporučuje se fizičko hlađenje tokom hipertermije (ventilacija, trljanje s alkoholom, ocat), uvođenje litičnih smjesa. Kod ponovljenog FS, preporučuje se da se roditelji nauče da injektiraju diazepam u dozi od 0.5-1.5 ml intramuskularno u vrijeme napada. To se radi kako bi se spriječio razvoj dugotrajnog napadaja i epileptičkog statusa. Terapija
AEP se ne sprovodi. U nekim slučajevima, moguće je profilaktičko davanje benzodiazepina ili fenobarbitala u terapijskoj dozi za period groznice (3-5 dana). Međutim, efikasnost ovakve "prevencije" nije dokazana.
U slučaju dijagnoze atipičnih FS, naprotiv, potrebno je propisati liječenje, kao kod epilepsije (na primjer, karbamazepina ili preparata valproata u doznim dozama). Kod teških dugotrajnih napada atipičnih FS, iste mjere se uzimaju kao i kod epileptičkog statusa.
Diferencijalna dijagnoza epilepsija se izvodi sa sinkopalnim stanjima, psihogenim poremećajima, poremećajima spavanja, neepileptičnim mioklonusom, migrenom, hiperkinezom. U kliničkoj praksi najveće poteškoće uzrokuje diferencijalna dijagnoza epilepsije sa sinkopalnim stanjima i konverzijskim (psihogenim) napadajima.
14.6. Opći principi liječenja epilepsije
Osnovni princip: maksimalna terapeutska efikasnost uz minimum nuspojava. Pacijenti koji pate od epilepsije primorani su da koriste AED dugi niz godina. U tom smislu, važan uslov za terapiju je odsustvo negativnih efekata lekova na kvalitet života pacijenata.
Pacijent mora da se pridržava sna i budnosti; izbegavajte dovoljno sna, kasno spavanje i rano (naročito iznenadno) buđenje. Adolescentima i odraslim pacijentima se savetuje da se uzdrže od pijenja. Treba izbegavati izlaganje ritmičkoj stimulaciji svetlosti kod oblika epilepsije sa teškom fotosenzitivnošću. Strogo pridržavanje ovih pravila može smanjiti učestalost epileptičkih napada kod 20% pacijenata.
Lečenje epilepsije može se započeti tek nakon što se ustanovi tačna dijagnoza. Preventivno lečenje epilepsije je neprihvatljivo! Promene u EEG u odsustvu bilo kakvih simptoma bolesti nisu razlog za propisivanje terapije. Izuzetak je teška traumatska povreda mozga (kontuzija mozga, pojava hematoma), nakon čega je moguće propisati osnovni AED za period od 6-12 mjeseci. Kod epileptičnih encefalopatija, terapija se može propisati u odsustvu napadaja na osnovu kombinacije difuzne epileptiformne aktivnosti sa izraženim poremećajima viših mentalnih funkcija.
Lečenje epilepsije treba početi nakon drugog napada. Jedan paroksizam može biti "slučajan", uzrokovan groznicom, metaboličkim poremećajima i ne odnosi se na epilepsiju. AEP se propisuje samo u slučaju ponovljenih neprovociranih epileptičkih napadaja. Kod nekih benignih epileptičkih sindroma djetinjstva (prvenstveno ER) i refleksnih oblika epilepsije (čitanje epilepsije, primarne fotosenzitivne epilepsije), pacijentima bez AED dozvoljeno je da se savladaju ako su napadaji vrlo rijetki i lako se javljaju tijekom preventivnih mjera.
Prilikom propisivanja AEP-a važno je uzeti u obzir princip monoterapije: započinjanje liječenja se provodi jednim lijekom. Monoterapija izbjegava pojavu teških nuspojava i teratogenih efekata, čija se učestalost značajno povećava uz istovremeno imenovanje nekoliko lijekova. Izuzetak su rezistentni oblici epilepsije (Westov sindrom, Lennox-Gastautov sindrom, simptomatska fokalna epilepsija), u kojem je nemoguće postići efekat bez upotrebe kombinirane terapije. Preparati se propisuju strogo u skladu sa oblikom epilepsije i prirodom napada. Uspeh lečenja epilepsije u velikoj meri je određen tačnošću sindromološke dijagnoze.
Prvi put se lijek propisuje, počevši od male doze, postepeno ga povećavajući dok se ne postigne terapijski efekat ili se pojave prvi znaci nuspojava. Ovo uzima u obzir djelotvornost i podnošljivost lijeka, a ne njegov sadržaj u krvi. Početna doza je obično 1 / 8-1 / 4 procijenjenog prosječnog terapeutika. Povećanje doze se javlja svakih 5-7 dana (u zavisnosti od tolerancije leka i obeležja epilepsije).
Potrebno je propisati AED u adekvatnim doznim godinama. Upotreba malih doza je jedan od glavnih razloga za “pseudorezistenciju” u kliničkoj praksi. Treba imati na umu da je kod teških oblika epilepsije jedina šansa da se pacijentu zaista pomogne da prepiše AED u visokim dozama (Tabela 24).
Ako je lek nedelotvoran, on se postepeno zamenjuje drugim, potencijalno efikasnim u ovom obliku epilepsije. Nemoguće je odmah dodati drugi lijek, odnosno prebaciti se na polieterapiju.
Postoji oko 30 AED-ova sa različitim spektrom anti-epileptičkih aktivnosti i sporednih efekata. Prednost se daje savremenom AED-u sa širokim spektrom kliničke efikasnosti i dobrim podnošenjem (valproat, topiramat). Preporučuje se tretman sa produženim lekovima koji se propisuju 2 puta dnevno (konjuks retard, depakin-hrono, retleps finlepsin, tegretol CR). Valproati (Depakin, Convulex, Convulsofine) i Karbamazepin (Finlepsin, Tegretol, Trileptal) se nazivaju osnovni AEP. Sukcinimidi (sukilep), benzodiazepini (klonazepam, klobazam) i lamotrigin (lamictal) se koriste u djece kao dodatna terapija. Novi AED (topiramat, levetiracetam, okskarbazepin) se propisuju kao monoterapija iu kombinaciji sa osnovnim AEP. Stari AEP uključuju barbiturate (fenobarbital, heksamidin, benzon) i hidantoine (difenin, fenitoin); imaju ozbiljne neželjene efekte i ređe se propisuju u poslednje vreme.
Za kontrolu terapije i nuspojava potrebno je obaviti krvnu analizu jednom u 3 mjeseca uz obavezno ispitivanje broja trombocita, kao i biokemijski test krvi sa određivanjem bilirubina, holesterola, jetrenih enzima. Svakih 6 mjeseci izvodi se ultrazvuk abdomena. Takođe se preporučuje praćenje nivoa antiepileptika u krvi tokom svakog pregleda. Vodite dnevnik pacijenta ili njegovih roditelja.
Tabela 24.Razlozi nedostatka efekta na imenovanje AED-a
U rezistentnim slučajevima ukazuju se na hiruršku resekciju, stimulaciju vagusnog živca i ketogenu dijetu. Potrebno je strogo se pridržavati indikacija za hirurško liječenje, pacijentima je potrebno provesti predkirurški pregled, koji je moguć samo u specijaliziranim centrima. S epizodama simptomatske fokalne epilepsije rezistentne na AED, kirurško liječenje može biti jedini način da se pacijenti u potpunosti oslobode napadaja.
Trajna remisija - bez napadaja 1 godinu ili duže. O potpunoj remisiji govore u odsustvu napada i normalizaciji EEG-a.
Uslovi redukcije i ukidanja AEP su strogo individualni i zavise prvenstveno od oblika epilepsije i karakteristika tijeka bolesti. U idiopatskim fokalnim oblicima i epilepsiji dječjeg apscesa, smanjenje AEP može početi nakon 3 godine bez napadaja; sa simptomatskom žarišnom epilepsijom, mladenačkim varijantama idiopatske generalizovane epilepsije - ne ranije od 4 godine remisije. Potpuno ukidanje AEP-a vrši se postepeno, obično u roku od 1 godine.
Faktori nepovoljne prognoze: prematurnost u anamnezi, rani početak napadaja, epileptički statusi, fokalni neurološki poremećaji, smanjena inteligencija, izraženi poremećaji ponašanja, prisustvo kontinuiranog regionalnog usporavanja na EEG ili fenomen sekundarne bilateralne sinhronizacije, nedostatak efekta primjene osnovnog AEP u adekvatne doze. Trenutno, zahvaljujući korišćenju celokupnog arsenala AEP-a, 65% pacijenata uspeva da postigne trajnu remisiju napada.
14.7. Status epilepticus
Status epilepticus (ES) je napad koji traje više od 30 minuta ili čestih čestih napada, između kojih svijest nije u potpunosti obnovljena. Države koje su ugrožene razvojem ES: dugi (više od 5 min) napad ili više od 3 generalizirana konvulzivna napadaja do kojih je došlo u roku od 24 sata.
U prosjeku, učestalost pojave ES je 28 slučajeva na 100 000 ukupne populacije i 41 na 100 000 dječje populacije. U 5% odraslih pacijenata i 20% djece sa epilepsijom, došlo je do istorije ES. U 26% slučajeva ES se javlja kod djece starosti 1 godine, u 43% slučajeva -
u prve 2 godine, au prve 3 godine - u 54%. ES čini do 4% svih slučajeva u hitnoj neurologiji. Smrtnost u ES u odsustvu specijalizovane nege je do 50%, a uz adekvatan tretman - 5-12%.
Pogoršanje tijeka epilepsije, nepravilno davanje AED, zarazne bolesti sa groznicom dovode do ES. ES otežavaju povrede glave, hematome, moždani udar, neuroinfekciju, egzogenu intoksikaciju, teške metaboličke poremećaje itd.
Patogeneza ES uključuje 2 faze.
1. Kontinuirana aktivnost napadaja ubrzava metabolizam mozga, kao odgovor na cerebralni protok krvi i povećanje kisika i glukoze. Postepeno, kompenzacijski mehanizmi su osiromašeni, razvija se acidoza, a nivo laktata u tkivu mozga se povećava. To dovodi do narušene srčane aktivnosti: povišenog krvnog pritiska, srčanog volumena i srčanog ritma. Povećanje aktivnosti simpatičkog sistema dovodi do pojave hipersalivacije, znojenja, povećanja bronhijalne sekrecije i hiperpireksije. Postoji hiperglikemija zbog povećanog oslobađanja adrenalina i noradrenalina.
2. Poremećaj kompenzacionih mehanizama dovodi do razvoja edema i distrofije neurona, što dodatno povećava epileptogenezu. Cerebralni protok krvi počinje da zavisi od sistemskog arterijskog pritiska. Hipoksija se produbljuje zbog hipoksije, neke lekovi (na primjer, intravenozno davanje Relaniuma). Razvijen je edem mozga, smanjen cerebralni protok krvi. Rezultat svih ovih promena je cerebralna ishemija, hipoksija i acidoza. Kasnije se javlja oštećenje više organa: sistemska acidoza, hipoglikemija, abnormalna funkcija jetre, zatajenje bubrega, rabdomioliza, DIC. Moguće su komplikacije intenzivne njege: infekcije, plućna embolija, neravnoteža elektrolita.
Tokom emitovanja ES:
Prostata (0-9 min od početka napada);
Početno (10-30 min);
Razmešteno (31-60 min);
Vatrostalni (preko 60 min).
Convulsive ES- Ovo je stanje u kojem perzistentni ili intermitentni toničko-klonični napadi traju duže od 30 minuta bez obnavljanja svijesti između napada. Konvulzivni ES čini 10-25% svih slučajeva ES. U početku, napadi postaju češći ili produženi (sa razvojem države kojoj prijeti ES). U ovom periodu može se spriječiti razvoj ES. Tipične toničko-klonične konvulzije vremenom postaju sve češće, dolazi do potpunog gubitka svijesti. U stanju kome, klonska aktivnost se može smanjiti - gotovo do potpunog nestanka. U ovom trenutku, respiratorni, cirkulatorni i metabolički poremećaji se povećavaju. Na EEG-u sa konvulzivnim ES, uočena je generalizovana epileptiformna aktivnost u obliku oštrih valova, šiljaka, kompleksa brzih valova, a zatim usporavanja. Bioelektrična aktivnost je maskirana velikim brojem miografskih i motoričkih artefakata. U drugoj fazi ES, glavna aktivnost usporava i poravnava se. Pojava “periodičnih lateraliziranih epileptiformnih poremećaja” i trifaznih valova se opaža s dugim trajanjem ES i označava nepovoljnu prognozu (smrt ili razvoj vegetativnog stanja). Smrtnost u konvulzivnim ES je 5-19% i zavisi od etiologije. Neurološki i mentalni poremećaji su proporcionalni trajanju statusa.
Poseban oblik ES kod djece je hemikonvulzivni hemiplegični epileptički sindrom.Pojavljuje se kod djece prve 4 godine života, češće sa vrućicom, a karakterizira je težak dugotrajan status generaliziranih konvulzivnih napada s izrazitim unilateralnim naglaskom. Nakon prestanka statusa pacijenta, javlja se produžena hemiplegija, koja se javlja na strani prevalencije napadaja. U većini slučajeva prognoza je loša - u budućnosti 85% djece razvija simptomatsko liječenje fokalna epilepsija sa intelektualnim invaliditetom i motornim deficitom.
ES za epilepsiju Kozhevnikov manifestuju se uporni mioklonični napadi, ograničeni na određeni segment tela. Povremeno se mogu pojaviti motorizovani Jackson napadi sa sekundarnom generalizacijom.
ES mioklonični napadaji manifestuju se nekontrolisane česte, gotovo kontinuirane mioklonije, izraženije u distalnim ekstremitetima, a prati ih
zapanjen, a ne potpuni gubitak svijesti. Mioklonični status epilepticus nastavlja se podmuklo, nastaje postepeno i može trajati nekoliko dana, mjeseci, pa čak i godina, praćeno progresivnom demencijom. EEG u miokloničkom statusu obično otkriva višestruka polispike-valna pražnjenja u odsustvu fiziološke pozadinske aktivnosti, kao i difuzno kontinuirano usporavanje naizmenično sa višestrukim multifokalnim šiljcima i difuznim i generalizovanim kompleksima spike i polispike.
Neuverljiv ES (status odsutnosti) karakteriše pojava regularne generalizirane aktivnosti vrha-vala na EEG-u. Najtipičniji tip odsutnosti u djetinjstvu je ES tipičnih izostanaka (stupor vršnog vala). Najčešće se uočava u okviru dječje i adolescentne absane epilepsije, rjeđe u adolescentnoj miokloničnoj epilepsiji. Ona se manifestuje oštrim povećanjem izostanaka, slijedeći jedan za drugim direktno ili u vrlo kratkom intervalu. Tu su amymia, drooling, stupor. Dijete izgleda sanjivo, pokreti su spori. Stepen poremećaja svijesti varira. Deca ponekad zadržavaju sposobnost da reaguju na grad i obavljaju jednostavne zadatke. Mioklonije mišića lica, ramena, ruku se mogu odrediti. Trajanje statusa je od nekoliko minuta do nekoliko sati pa čak i dana. Status izostanaka se često javlja ujutro, odmah nakon što se pacijenti probude i često završava generaliziranim konvulzivnim napadom. U polovini pacijenata, status se ponavlja sa neadekvatnim tretmanom. Status odsustva izazvan je nedostatkom sna ili nepravilnim tretmanom, posebno upotrebom karbamazepina i vigabatrina.
ES kompleksni fokalni napadaji klinički promenljiva. Obično počinje periodom zbunjene svesti, koji traje od nekoliko sati do nekoliko dana. Oči širom otvorene, lice hipomimično. Postoje dugoročni ambulantni automatizmi, koji liče na svrsishodne, svrsishodne i koordinirane pokrete, obično sa interakcijom. U ovom stanju pacijenti mogu besciljno lutati ulicama; Sedite u prevoz, idite u druge gradove. Često, svijest nije potpuno isključena, pa se može održati djelomičan kontakt s pacijentom. Mogući predisponirajući faktori su unos alkohola, ovisnost o drogama, infekcije, menstruacija, elektro-elek- tiv
horny therapy. Na EEG-u, konstantna ili periodična, regionalna (najčešće u temporalnim tragovima), paroksizmalna aktivnost je uočena u obliku kompleksa vršnih talasa, izolovanih pikova ili usporene aktivnosti vršnog vala.
Liječenje epileptičkog statusa. U početnim fazama, preporučljivo je koristiti lijekove brzog djelovanja, au kasnijim fazama lijekove koji se ne akumuliraju u tijelu i imaju najmanje nuspojava. Terapijske mere za ES su strogo diferencirane u zavisnosti od stadijuma ES: u 1. fazi terapijske mere se sprovode u pretpozicionom periodu; u drugom i trećem - u uslovima jedinice za intenzivnu negu neurološkog odeljenja u četvrtom - u jedinici intenzivne nege. U drugoj fazi potrebno je sprovesti sve dijagnostičke mjere za identifikaciju etiologije ES i pratiti vitalne znakove.
I. Pre status (0-9 min od početka napada) - pravovremeno započeti adekvatan tretman može spriječiti razvoj teških ES:
Osiguravanje dišnih puteva;
Terapija kisikom;
Diazepam (u 2 ml od 10 mg) / u 0,25 mg / kg, brzina davanja - 2-4 mg / min. Možda se ponavlja svakih 30 min. Ukupna doza lijeka dnevno ne smije prelaziti 40 mg. Glavni sporedni efekat je respiratorna depresija.
Ii. Rani status (10-30 min):
Nastavi diazepam;
Lorazepam (u 1 ml 4 mg) 0,05-0,1 mg / kg pri brzini od 2 mg / min. Daje se 1 ili 2 puta u intervalu od 20 minuta, ukupno - ne više od 4 mg. Nuspojave: razvoj tolerancije nakon 1-2 injekcije; rijetko, respiratorna depresija (manje izražena nego kod diazepama), hipotenzija;
Fenitoin (difantoin) (u 5 ml od 250 mg) u / u, razblažen u fiziološkoj otopini 5-20 mg / ml. Doziranje - 15-20 mg / kg pri brzini od 25 mg / min. Moguće je ponovo davati lek svakih 6 sati u dozi od 5 mg / kg IV ili oralno putem sonde. Koncentracija fenitoina u krvi treba da se održava na nivou od 20-25 μg / ml.
Nuspojave: zastoj srca, hipotenzija, fleboskleroza. U odsustvu fenitoina, moguće je primijeniti natrijev oksibutirat (GHB) (u 1 ml 20% otopine od 200 mg) IV. Doziranje - 100-150 mg / kg pri brzini od 400 mg / min. Nuspojava je hipokalemija.
Iii. Prošireni status (31-60 min):
Diazepam ili lorazepam;
Fenobarbital (u 1 ml 200 mg) IV. Doziranje djece mlađe od 1 godine - 20 mg / kg, zatim - 15 mg / kg pri brzini do 100 mg / min. Pojedinačna doza ne smije prelaziti maksimalnu starost ili biti veća od 1000 mg. Dozu je moguće davati svakih 8 sati u dozi od 3-5 mg / kg / dan oralno kroz epruvetu. Nuspojave: smanjenje kontraktilnosti miokarda, depresija disanja, depresija svijesti, arterijska hipotenzija;
Alternativa je intravenska injekcija depakina u dozi od 20-25 mg / kg tokom prvih 5-10 minuta, zatim 2 mg / kg / h. Standardna doza je 25 mg / kg / dan. Održava se doza od 5 mg 4 puta dnevno ili se vrši kontinuirana infuzija od 1 mg / kg / h. Depakine za intravenozno davanje ne inhibira disanje i srčanu aktivnost; potrebna je koncentracija u krvnoj plazmi; omogućava vam da izbegnete intubaciju pacijenta; visoko efikasan (80-90%), uključujući neefikasnost diazepama i fenitoina; ne garantuje ponavljanje napada u roku od 24 sata.
Iv. Refraktorni status (preko 60 min) praćen je teškim, često ireverzibilnim promjenama u mozgu i unutrašnji organiMetabolički poremećaji:
Intubacija pacijenta sa prelaskom na veštačku ventilaciju pluća u jedinici intenzivne nege;
Barbiturna anestezija: uvođenje natrijum tiopental (u 1 ml 2,5% rastvora od 25 mg) / u prosečnoj dozi od 100-250 mg tokom 20 sekundi. U odsustvu efekta - dodatna primjena lijeka u dozi od 50 mg u / v svaka 3 minute do potpunog oslobađanja od napadaja. Zatim, prelazak na dozu održavanja - u proseku 3-5 mg / kg i / h svakih sat vremena (zahteva stalno praćenje leka u krvi). Ukupna doza lijeka ne smije prelaziti 1 g. Trajanje barbiturne anestezije je obično 12-24 sata, komplikacije: smanjenje kontraktilnosti miokarda, teška respiratorna depresija,
hipotenzija, toksični hepatitis i pankreatitis, anafilaktički šok;
Nakon eliminacije ES i obnove svijesti - prelazak na oralnu primjenu neophodnih antiepileptičkih lijekova.
Tokom faze 2-4 ES, sprovodi se dodatna terapija u cilju korekcije vitalnih funkcija, elektrolitski poremećajiboriti se protiv cerebralnog edema (natrijeva sol deksametazona 4 mg i.v. svakih 6 sati ili manitol 1,0-1,5 g / kg i.v. u kapanju brzinom od 60-80 kapi / min).
Za lečenje epileptičkog statusa kod dece od 1 godine starosti primeni:
Benzodiazepini
Diazepam (0,5 mg / kg po rektumu, IM ili IV),
Lorazepam (0,2 mg / kg po rektumu ili IV)
Midazolam (0.15-0.4 mg / kg i.v. bolus, koji podržava infuziju - 1-3 mcg / kg / min);
Hydantoins
Phosphenitoin (20 mg / kg i.v.)
Fenitoin (20 mg / kg i.v., maksimalna brzina davanja je 25 mg / min, koncentracija u plazmi je 20-25 μg / ml).
Sa vatrostalnim statusom primeni:
Natrijum hidroksibutirat (GHB) u dozi od 100-150 mg / kg pri brzini od 400 mg / min;
Fenobarbital (20 mg / kg IV, re-bolus nakon 20-30 minuta, maksimalna doza - 100 mg / kg dnevno);
Propofol (3 mg / kg IV bolus, zatim infuzija od 100 µg / kg / min).
Injekcijski depakin 400 mg u 1 bočici: početna doza - 15-25 mg / kg, zatim infuzija za održavanje - 1-4 mg / kg / h.
Prognoza.Ishodi ES mogu biti: potpuni oporavak, oporavak od prisustva upornih povreda, smrt. Općenito, rizik od razvoja različitih komplikacija je veći, što je dijete mlađe. Mnogi pacijenti nakon konvulzivne ES na CT ili MRI otkrivaju difuznu ili lokalnu atrofiju moždane kore. Prije korištenja modernih metoda liječenja u ranom dvadesetom stoljeću. mortalitet od ES iznosio je 51%; krajem dvadesetog veka. -18%. Smrtnost je veća kod ES na osnovu moždanog udara, encefalitisa, traumatskih povreda mozga i tumora mozga. Letalni ishod je izuzetno rijedak u ES u okviru idiopatske generalizirane epilepsije (ne više od 1% slučajeva).
1. Karlov V.A.Konvulzivni epileptički status: riješen i nerešen // Neurološki časopis. - 2000. -? 3. - str.
2. Litvinovich EF, Savchenko A.Yu., Pospolit A.V.Savremeni aspekti farmakoterapije epileptičkog statusa // Klinička epileptologija. - 2007. -? 1. - str.
3. Mukhin K.Yu., Petrukhin A.S.Idiopatski oblici epilepsije: sistematika, dijagnoza, terapija. - M .: Medicina, 2000. -
319 s.
4. Petrukhin A.S.Epileptologija djetinjstva. - M.: Medicina,
2000. - 623 str.
5. Aicardi J., Chevrie J.J.Konvulzivni status epileptika kod dojenčadi i djece // Epilepsija. - 1970. - Vol. 11. - R. 187-197.
6. Aminoff M.J., Simon R.P.Status epilepticus: uzroci, klinička obilježja i posljedice kod 98 pacijenata // Am. J. Med. - 1980. - Vol. 69. -
R. 657-666.
7. Brown J.K., Hussain N.H.Status epilepticus 1: patogeneza // Dev. Med. Clin. Neurol. - 1991. - Vol. 33. - P. 3-17.
8. Cascino G.D., Hesdorffer D., Logroscino G., Hauser W.A.Morbiditet nonfebrile status epilepticus u Rochesteru, Minnesota, 1965-1984 //
Epilepsija. - 1998. - Vol. 39/8. - P. 829-832.
9. Cockerel O.C., Walker S.M., Sander J.W., Shorvon S.D.Kompleksno parcijalno
status epilepticus: povratni problem // J. Neurol. Neurosurg Psychiatry. -
1994. - Vol. 57. - P. 835-837.
10. DeLorenzo R.J., Garnett L.K., Towne A.R. et al.Poređenje epileptika sa produženim epizodama napadaja od 10 do 29 minuta //
Epilepsija. - 1999. - Vol. 40/2. - P. 164-169.
11. Philips S.A., Shanahan R.J.Etiologija i mortalitet epileptika kod djece // Arch Neurol. - 1989. - Vol. 46 - 74-76.
12. Sander J.W.A.S., Hart Y.M., Trevisol-Bittencourt P.S.Absense status //
Neurologija. - 1990. - Vol. 40. - P. 1010.
13. Shorvon S.D.Status epilepticus: - Cambridge: Cambridge University Press, 1994.
14. Wasterlain C.G. i Treiman D.M.Status Epilepticus: mehanizmi i upravljanje. - London: The MIT Press, 2006.
15. Treiman D.M.Status epilepticus // U: Udžbenik epilepsije. - Eds. J. Laidlaw, A. Richens, D. Chadwick. - Edinburg: Churchill-Livingstone,
1993. - P. 205-220.
Prema definiciji Međunarodne antiepileptičke lige (International League Against Epilepsy - ILAE) 2005, epileptički napad je prolazna klinička manifestacija patološke prekomjerne ili sinkrone neuronske aktivnosti mozga. 
Da biste pravilno dijagnosticirali epilepsiju, prvo morate utvrditi vrstu epileptičnog napadaja u skladu sa savremenom međunarodnom klasifikacijom epileptičkih napadaja, koristeći novu definiciju termina "epilepsija".
Prva faza dijagnoze je prikupljanje informacija o samom napadu, njegovoj fenomenologiji, vjerovatnoći njene provokacije; optimalno sa video snimkom samog napada.
Druga faza dijagnoze - nakon utvrđivanja činjenice epileptičkog napada, potrebno je utvrditi njen tip, prema klasifikaciji. Godine 1981. usvojena je klasifikacija epileptičkih napadaja, ali diskusije o njenom poboljšanju se nastavljaju. U 2016. godini predstavljena je ažurirana radna klasifikacija epileptičkih napadaja, koja se može koristiti u praksi, ali će biti konačno usvojena kasnije, a očekuje se 2017.
Klasifikacija epileptičkih napadaja (ILAE, 2016), osnovna šema:
1. Fokalna:
- motor;
- podvozje;
- bilateralni tonik-klon.
2. Generalizovano:
- motor;
- odsustva.
3. Sa nepoznatim početkom:
- motor;
- hod
4. Neklasificirano.
Za sve napade potrebno je ukazati na nivo oštećenja svijesti: napad bez oštećenja svijesti, sa oslabljenom sviješću, sa nepoznatom sviješću.
Klasifikacija fokalnih epileptičkih napadaja (ILAE, 2016):
1. Motor:
- tonik;
- atonic;
- mioklonska;
- clonic;
- epileptički spazam;
- hypermotor
2. trčanje:
- senzorna;
- kognitivne (halucinacije, deja vu, iluzije, oslabljena pažnja, afazija, opsesivne misli)
emocionalno (uznemirenost, agresija, suza, smijeh) - vegetativni (brady-, tahikardija, asistolija, osjećaj hladnoće ili toplote, crvenilo ili bljedilo kože, gastrointestinalni poremećaji, groznica, hiper, hipoventilacija, mučnina, povraćanje, piloerekcija, itd.)
- automatizmi (agresija, ručno (u rukama), orofacijalni, seksualni, vokalizacijski, složeni pokreti u obliku hodanja ili jogginga, svlačenja).
3. Bilateralni tonik-klon (u poslednjoj klasifikaciji - sa sekundarnom generalizacijom).
Za fokalne napade, za svaki tip napada, potrebno je zapaziti stepen svijesti: napad bez poremećaja svijesti, s kršenjem svijesti, s nepoznatom sviješću.
U 2016. godini, ILAE je izvršio neke izmjene u terminologiji napada. Dakle, preporučuje se da se termin „parcijalni“ napadi zameni „fokalnim“ (sa / bez poremećaja svesti i sa nepoznatom svešću), „kompleksnim parcijalnim“ napadima - sa „žarištem sa oslabljenom svešću“.
Gotovo 60% epileptičkih napada je lokalno i samo 23% su generalizirani tonik-klon. Prema savremenim istraživanjima, epilepsija je sistemske bolesti mozak povezan sa kršenjem neuronskih veza, a ne samo lokalna disfunkcija mozga. Pri privlačenju mnogih neuronskih veza, epileptički napadaji mogu nastati iz neokortikalnih, thalamo-kortikalnih, limbičkih i stabljičkih sekcija.
Treća faza dijagnoze. Pored utvrđivanja vrste napada, neophodno je sprovesti i aktuelnu dijagnozu napada, odnosno utvrditi lokaciju epileptičkog fokusa, ako su ovi napadi lokalni. Napadi koji nastaju zbog prekomerne patološke pobude određene grupe neurona u različitim režnjevima mozga imaju svoje karakteristike.
Vremenski epileptični napadi: dnevni, javljaju se s učestalošću od nekoliko puta mjesečno, rijetko komplicirani epileptičkim statusom, manifestne manifestne pojave - često s aurom (vegetativno, psihotično, često s oslabljenom sviješću, automatizmom (oralni, verbalni), motorni fenomeni su tekući, mogu biti distonične instalacije, postiktalni poremećaji svijesti.
Frontalni epileptički napadaji imaju sledeće karakteristike: česte klasterske napade, kratkotrajni protok (20-40 sekundi), često razvoj u snu, često sa sekundarnom generalizacijom na epistatičku struju, polimorfne aure sa naglim početkom, rane debi dominantne motorne promene - pareza, paraliza, disografija itd., mogu da nastave sa poremećajima svesti, oporavkom nakon napada brzo. Tonični frontalni napadi najčešće se dijagnosticiraju (oko 64%), zatim klonični (36%) i epileptički spazmi (36%).
Za fokalne epileptičke napadaje sa žarištima u zadnjem koritu, vizuelne, somatosenzorne, vegetativne, gustatorne aure, karakteristične su neželjene napade i opsoklonus oka, treptanje, anosognozija, akaculija, apraksija, aleksija.
Četvrta faza dijagnoze. Vrste epileptičkih napadaja su osnova za uspostavljanje oblika epilepsije, prema klasifikaciji iz 1989. Prema definiciji iz 2005. godine, epilepsija je poremećaj mozga koji se odlikuje stalnom sklonošću epileptičkim napadima, kao i neurobiološkim, pozitivnim, psihološkim i socijalnim posljedicama ovog stanja. Ova definicija epilepsije uključuje razvoj najmanje jednog epileptičkog napada. Termin "poremećaj" nije dovoljno jasan pacijentima i ozbiljnost stanja, stoga su ILAE i Međunarodni biro za epilepsiju (IBE) nedavno donijeli zajedničku odluku o liječenju epilepsije kao bolesti. U 2014. godini usvojena je nova praktična definicija epilepsije, prema kojoj je epilepsija bolest mozga koja odgovara na slijedeća stanja:
- najmanje dva neprovocirana ili refleksna epileptička napadaja sa intervalom od najmanje 24 sata;
- jedan neprovocirani (refleksni) epileptički napad i vjerovatnoća ponovljenih napadaja, što odgovara ukupnom riziku od recidiva (\u003e 60%) nakon dva neprovocirana napadaja u narednih 10 godina
- dijagnoza epileptičkog sindroma (npr. Westov sindrom).
Kriterijumi za "završetak" epilepsije uključuju postizanje određene starosti kod pacijenata sa oblikom epilepsije, u zavisnosti od starosti, ili odsustvo epileptičkih napadaja tokom 10 godina kod pacijenata koji nisu primali antikonvulzivne lekove duže od 5 godina. Radna grupa je radila na terminu "izlečenje", što ukazuje da rizik od epileptičkih napadaja nije veći od rizika od \\ t zdravi ljudimeđutim, kod pacijenata sa epilepsijom u istoriji ovakav nizak rizik nikada nije postignut. Termin "remisija" nije dobro shvaćen i ne ukazuje na odsustvo bolesti. Radna grupa je predložila termin “završetak” epilepsije, što ukazuje da pacijent nema epilepsiju, ali je nemoguće isključiti pojavu napadaja u budućnosti. Rizik od rekurentnih napadaja zavisi od oblika epilepsije, starosti, etiologije, tretmana i drugih faktora. Na primjer, u juvenilnoj miokloničnoj epilepsiji, rizik od rekurentnih napadaja ostaje visok desetljećima. Strukturne lezije mozga, kongenitalne defekte praćene su stalnom sklonošću epileptičkim napadima. U studiji koja je obuhvatila 347 djece bez napadaja najmanje 5 godina (bez uzimanja antikonvulzivnih lijekova), kod 6% djece zabilježeni su kasni recidivi.
Uzroci epilepsije
Više od polovine djece s epilepsijom pati od idiopatskog oblika bolesti u kojem nema drugih utvrđenih uzroka od genetskih. U klasifikaciji AT Berg i koautori (2010), umjesto izraza "idiopatski", predlaže se "genetski", što je rezultat već poznatih i sumnjivih gena. Mnogi geni su već poznati (autosomno dominantna noćna frontalna epilepsija, itd.).
Epilepsija, čiji je uzrok poznat i nije povezan sa genetskim faktorima, naziva se simptomatska (strukturalna / metabolička), u terminologiji AT Berg et al. (2010). U ovom slučaju, epilepsija je sekundarni rezultat određene utvrđene strukturne ili metaboličke bolesti:
- oštećenje mozga zbog hronične hipoksije i asfiksije rađanja, trauma rođenja, subduralne hematome, kongenitalne TORCH infekcije;
- metaboličke bolesti (metabolički poremećaji aminokiselina, ugljikohidrata, itd.), koji su popraćeni multiorganskim simptomima, osim epilepsije;
mitohondrijske bolesti; - kongenitalne malformacije mozga;
- hromozomski sindromi: Angelman, Down sindrom, krhki X hromozom, itd.;
- nasljedni neurotični sindromi (Phakomatozy): tubularna skleroza, itd.;
- traumatska povreda mozga;
- vaskularne arteriovenske malformacije mozga;
- pretrpio moždani udar.
Postoji još jedna grupa epilepsije sa nepoznatom etiologijom (ranije nazvana kriptogena epilepsija), čiji uzrok još nije utvrđen, može biti genetski ili strukturno-metabolički.
Prognoza zavisi od obima i uzroka oštećenja mozga. Dakle, teške prenatalne lezije mogu biti teško liječiti.
Međunarodna klasifikacija epilepsija i epileptičkih sindroma ILAE 1989 (skraćena verzija)
Lokalizacija (fokalna, parcijalna) epilepsija i sindromi
1. Idiopatski (genetski):
- benigna epilepsija djetinjstva sa centralnim temporalnim adhezijama na elektroencefalogramu (EEG) (rolandska)
- benigna pedijatrijska epilepsija sa zatiljnim napadima (Gasto sindrom)
- benigna parcijalna ocipitalna epilepsija sa ranim debutom (Panayotopoulos sindrom)
primarna epilepsija kod čitanja; - autosomno dominantna frontalna epilepsija.
2. Simptomatski (strukturni / metabolički):
- hronična progresivna parcijalna epilepsija djetinjstva (Kozhevnikova)
Rasmussenov sindrom; - epilepsija, koju karakterišu napadaji uzrokovani specifičnim precipitacionim faktorima;
- epilepsija temporalnog režnja;
- frontalna epilepsija;
- parijetalna epilepsija;
- okcipitalna epilepsija.
3. Kriptogeno (nepoznato).
1. Idiopatska (genetska) epilepsija
Benigna epilepsija u detinjstvu sa centro temporalnim šiljcima na EEG (rolandska epilepsija)
Učestalost u populaciji je 21 na 100 hiljada.
Dijagnostikuje se kod 15-25% sve djece školskog uzrasta s epilepsijom. Bolest debituje u dobi od 4 do 10 godina sa maksimalno 9 godina. Dečaci su češće bolesni nego djevojčice. Klinički se manifestuje karakteristične znakove: počevši od senzomotorne aure, pojavljuju se "grlo" zvuci ili anarthria, hemifacijalni kratki motorni napadi noću kada zaspavaju i budi se, u 20% - također i faciesbrachial grčevi, u 25% slučajeva na otvaranju posmatraju sekundarne generalizirane napadaje. Trajanje napada: jednostavno - 30-60 sekundi, sekundarno generalizovano - do 1-2 minuta sa učestalošću napada 2-6 puta godišnje (u dobi do 6 godina u početku bolesti - česti napadi). Ovaj oblik je benigni, tj. Osim epileptičkih napadaja, nema promjena u neurološkom statusu, kognitivnoj sferi - dijete može učiti u srednjoj školi. Bolest ima benigni tok; remisija se obično javlja kod 98% pacijenata prije puberteta.
Epileptiformne promjene između napada u 90% slučajeva;
tipično: benigne epileptiformne promjene u djetinjstvu (DEZD) u centralno-temporalnim vodovima (kao QRST na EKG-u), ali u dobi od 3-5 godina - u stražnje-okcipitalnim vodovima;
kod 30% djece se bilježe samo noćni EEG fenomeni (tijekom sporog spavanja - kompleksi vršnog vala). EEG normalizacija se javlja mnogo kasnije od kliničke remisije.
U lečenju se koristi samo monoterapija sa jednim od lekova prve linije: valproična kiselina, karbamazepin, lamotrigin, okskarbazepin, gabapentin, topiramat, levetiracetam. Međutim, postoje dokazi o mogućoj sekundarnoj bilateralnoj sinhronizaciji, posebno sa karbamazepinom i okskarbazepinom.
Benigna parcijalna ocipitalna epilepsija sa ranim debutom (Panayotopoulos sindrom)
Napadi se javljaju retko (do 5-7 u toku života), uglavnom tokom sna, manifestuju se odstupanjem očiju u stranu, narušenom sviješću o vrsti dezorijentacije, aktivnom povraćanju, nakon čega dolazi do napada glavobolje. U polovini djece napadaji se mogu produžiti - nekoliko sati sa gubitkom svijesti (iktalus i nesvjestica), praćeni povraćanjem, devijacijom očiju, kloničkim hemisudomima i postiktalnom glavoboljom.
Pedijatrijska okcipitalna epilepsija sa kasnim debutom (Gastho sindrom)
Napadi se evidentiraju češće nego kod Panayotopulos sindroma (1 put nedeljno - 1 put mesečno). Bolest počinje u dobi od 3-15 godina, sa maksimalno 8 godina. Klinička jezgra - jednostavni parcijalni senzorni napadi - vizuelne halucinacije u perifernom vidnom polju, hemianoptičke halucinacije, iluzije sa osećajem bola u očima, treptanjem, okretanjem očiju i glavom u suprotnom smjeru od epileptičkog fokusa. Trajanje napada je od sekunde do minuta. Na kraju napada karakteristične su pritužbe na jaku glavobolju sa povraćanjem (kod 50% pacijenata). Može postojati sekundarna generalizacija s toničko-kloničkim konvulzijama. U Panayotopoulos i Gasto sindromima nema promjena u procjeni neurološkog statusa i kognitivne sfere djeteta.
- DEZD u okcipitalnoj vodi kod 90% pacijenata između napada;
- glavna pozadina nepromijenjena;
- 30% djece može imati promjene u temporalnim tragovima;
- tipično: nestanak patološkog uzorka pri otvaranju očiju visokom fotosenzitivnošću;
- noćni EEG-video nadzor: u fazi sporog sna - povećanje DEZD-kompleksa ( rana dijagnoza bolesti) normalizaciju EEG-slike do dobi od 15 godina.
U tretmanu se primenjuje princip MONOTERAPIJE jednim od sledećih lekova - karbamazepin, preparati valproične kiseline, okskarbazepin, topiramat, lamotrigin.
Ovi oblici epilepsije se takođe smatraju benignim. Potpuna remisija u Panayotopoulos sindromu javlja se prije 9 godina, a kod Gastó sindroma 15 godina.
Autosomno dominantna frontalna epilepsija
Geni CHRNA4, CHRNA2 i CHRNB2 su lokalizirani na lokusima 20q13, 8q, 1p21. Ovaj oblik idiopatske epilepsije počinje češće u dobi od 7-12 godina. Odlikuje se noćnim napadima (nakon zaspanja, 2-3 sata prije buđenja). Početak se dešava sa vokalizacijom (obično vrisak), sa otvorenim očima. Po prirodi napada jednostavna i kompleksna parcijalna.
Polimorfizam klinike napada karakterističan je - složeni motorički činovi: dijete sjeda, grebu nos, glavu, pravi grimase, žvače pokret, dobija na sve četiri, ljulja, stvara pedaluvale ili boksne pokrete. U 70% slučajeva može biti aure (neugodni zvukovi, generalizovana jeza, vrtoglavica) - dijete se budi. Trajanje napada - do 1 min. Preko noći može biti više napada. U ovom obliku epilepsije postoji tendencija ka serijalnosti i „jakom jazu“ (nema napada za 2-3 meseca). Istraživanje ne otkriva promjene u neurološkom statusu, inteligenciji i govoru.
EEG karakteristike:
- glavna pozadina je nepromijenjena;
- sposoban za spavanje bez epileptičkih fenomena;
- glavna dijagnostička tehnika je noćni EEG video nadzor, pri čemu se regionalna aktivnost bilježi u frontalnim, frontalno-temporalnim vodovima.
Terapija je složena, obično efikasna polieterapija: karbamazepin, valproična kiselina, topiramat, lamotrigin, levetiracetam ili kombinacija osnovnih lijekova.
Ovaj oblik epilepsije zahtijeva diferencijalnu dijagnozu sa simptomatskom frontalnom epilepsijom, pri čemu EEG usporava osnovni ritam, neurološki status - bez fokalnih promjena, uz neuroizmanje - organske promjene u supstanciji mozga. Diferencijalna dijagnoza bi takođe trebala biti napravljena od parasomnija u kojima ne postoje epileptički obrasci na EEG-u.
2. Simptomatska (strukturna / metabolička) epilepsija
Frontalna epilepsija
Od svih simptomatskih i vjerovatno simptomatskih (kriptogenih) epilepsija, simptomatska frontalna epilepsija je 20%. Može početi u bilo kojoj dobi, u zavisnosti od uzroka. U zavisnosti od lokalizacije epileptogenog fenomena, izdvaja se 7 oblika frontalne epilepsije, a svaka se manifestuje svojim tipovima napada. Generalno karakterišu lokalni ili kompleksni napadaji koji se javljaju u frontalnom korteksu - kontralateralne klonične konvulzije, jednostruke, bilateralne tonične konvulzije koje završavaju Toddovom paralizom, složeni automatizmi koji izgledaju kao pokreti udova udova, ljuljanje tijela, pedaluvialni pokreti nogu. Epileptička pražnjenja u dodatnom frontalnom motornom prostoru manifestuju se kompleksnim fokalnim napadima u obliku toničkih konvulzija ruku, klasičnim "mačevalačkim položajem", adveracijom glave, bilateralnim proširenjem tijela, vrata, vokalizacijom. Aktivnost u području okretanja glave i očiju pokazuje adverziju očiju u suprotnom smjeru, trepćući. Svest se čuva ili nije potpuno izgubljena. Napadi sa fokusom u centralnoj zoni (područje korteksa u brazdi Rolanda) karakterišu Jacksonske marševe ili strogo lokalizovane klonske ili toničke konvulzije, konvulzije lica, gubitak mišićnog tonusa. Ako je koža nadražena, može doći do motornog napada bez narušavanja svijesti, grčeva lica sa gutanjem, pokretima za žvakanje, salivacije sa osećajem drugačijeg ukusa i grlenih simptoma. Napada noću, vrlo često, kratkoročno.
U neurološkom statusu otkrivaju se pareza, ataksija, intelektualni i govorni poremećaji.
EEG karakteristike:
- glavna pozadinska aktivnost je usporena;
- regionalna antiaktivnost (oštri valovi, oštro-sporokompleksi, vršni talasi)
bifrontalna ili difuzna aktivnost; - sekundarna bilateralna sinhronizacija (znak pogoršanja bolesti, pojava kognitivnog oštećenja).
Tretman je komplikovan. Vrlo često napadi otporni na adekvatnu terapiju. Neophodno je početi sa monoterapijom lijekovima prve linije u adekvatnoj dozi, a zatim preći na kombinaciju lijekova s različitim mehanizmima djelovanja, prema Protokolu za jedinstvenu terapiju epilepsije kod djece iz prve linije lijeka - karbamazepin (sa sekundarnom bilateralnom sinkronizacijom kontraindikovan), okskarbazepin, topiramat, drugi valproična kiselina, lamotrigin, treća - kombinacija lijekova.
Privremena epilepsija
Česti oblik svih simptomatskih epilepsija (30-35%). Debi se slavi u različitim godinama (obično u školi). Uobičajeni uzroci: efekti hipoksično-ishemijske encefalopatije u obliku glioze, kongenitalne malformacije (kortikalna displazija), arahnoidne ciste, efekti prenesenog encefalitisa, formiranje hipokampalne skleroze. Napadi mogu biti kod jednog pacijenta sa / bez oslabljene svesti. Dugi napadi - 1-2 minuta. Vegetativne manifestacije, mentalni i senzorni simptomi prisutni tokom cijelog napada ili samo na početku u obliku aure, fokalni napad nastavlja s oslabljenom svijesti s bilateralnim toničko-kloničkim napadima. Postoje dva oblika temporalne epilepsije, u zavisnosti od epileptogenog fokusa: medijalna (amigdala-hipokampalna) i lateralna (neokortikalna) epilepsija.
Medijalna (amigdala-hipokampalna) epilepsija je odgovorna za 65% svih temporalnih epilepsija i zbog prisustva fokusa u srednjim predjelima temporalnog režnja. Razlog je hipokampalna atrofija, često kod pacijenata kod kojih je došlo do složenih febrilnih napadaja prije dobi od 3 godine, posebno kod dugotrajnih unilateralnih napada (u 40% slučajeva). Nakon perioda remisije od 5-6 godina počinju fokalni česti rezistentni napadi, odnosno razvija se hronična epilepsija.
Klinička osnova ovog podtipa epilepsije:
- fokalni napadaji bez poremećaja svijesti - izolirana aura (vegetovisceralna, olfaktorna i ukusna halucinacija), mentalni fenomeni - stanje spavanja, depersonalizacija, derealizacija, strah, afekt, radosti, mentalni algoritmi sa očuvanom sviješću, distonijski položaj kontralateralne ruke, može biti u ipsilateralnoj ruci automatizmi;
- fokalni napadaji sa izolovanom deaktivacijom svijesti i automatizmom bez napadaja (dijalektički napadi).
Lateralna (neokortikalna) epilepsija karakteriše:
- slušne halucinacije
- vizuelne svijetle halucinacije (panoramski pogledi)
- autonomni napadaji (ne-sistemska vrtoglavica, “temporalna sinkopa” - spori pad bez ispitivanja sa montažom na distonski limb, automatizam)
- paroksizmalna senzorna afazija.
Pored čestih napada, sa brzim fokalnim promjenama u mozgu, djeca imaju neurološki deficit, kontralateralni fokus (parezu), emocionalne i intelektualne poremećaje.
EEG karakteristike:
- 50% pacijenata ima normalan EEG obrazac između napada;
potreban standard istraživanja - invazivne elektrode; - 30% pacijenata ima epitapiju između napada;
- sa medijalnom epilepsijom - promene u antero-kvadratnim vodovima;
- EEG žarišta ne mogu da se poklapaju sa morfološkim fokusom na magnetnu rezonancu (MRI) - formiranje "ogledalnog" fokusa;
- karakterističan EEG fenomen u otvaranju - nastavak regionalnog usporavanja aktivnosti;
- provokacija - ponekad nedostatak sna;
- noćni EEG ukazuje na 65% promjena između napada.
Na interiktalnom EEG - prednjem temporalnom fokusu adhezije, paroksizmalni theta ritam.
Karakteristike promena na MRI mozga u medijalnoj epilepsiji - atrofija hipokampusa, pojačani signal na T 2 iz hipokampusa. Hipokampalna skleroza napreduje.
Hirurško liječenje. Prognoza nakon hirurškog liječenja je dobra. Drug treatment kompleksan i ne uvek efikasan; često koriste polieterapiju.
Parijetalna epilepsija
Napadi su subjektivni, pa ih je teško detektovati, posebno kod male djece. Karakteristični somatosenzorni napadi u obliku osetljivog marsa u Jacksonu, često povezani sa motoričkim fenomenima. Somatosenzorni simptomi mogu biti pozitivni i negativni, mogu biti bolovi u trbuhu, mučnina, iluzija kretanja, nedostatak tjelesnog osjećaja (asmatognoza), vrtoglavica, dezorijentacija u prostoru. Može se razviti poremećena percepcija i govor (sa učešćem dominantne hemisfere), posturalnim ili rotacionim pokretima, sa širenjem impulsa, vizualnim simptomima (okcipitalno-temporalni parietalni), kontralateralni ili ipsilateralni pokreti sa distoničnim položajem udova u suprotnom smjeru ili prema uključenoj hemisferi. Vizuelne iluzije (makropsija, mikropsija, metamorfoza) ukazuju na prisustvo pražnjenja u stražnjim dijelovima parijetalne korteksa i parijetnoporoskopskih režnjeva.
Occipital Epilepsy
Kod 5% djece evidentiraju se svi simptomatski i kriptogeni oblici.
Pojavljuju se epileptička pražnjenja u primarnom vidnom korteksu:
- okulomotorni poremećaji (nistagmus, devijacija očiju u suprotnom smeru, bilateralna mioza)
- lokalni napadi bez poremećaja svijesti u vidu vizualnih halucinacija, iluzija, paroksizmalne amauroze, sužavanje vidnih polja;
- lokalni napadi sa oslabljenom sviješću i bilateralni toničko-klonički napadi;
- autonomni poremećaji kraja napada (glavobolja, povraćanje)
- acalculia, apraxia.
- Neurološki nedostatak zavisi od uzroka epilepsije. Često se javljaju okulomotorni poremećaji (poremećaj konvergencije, strabizam).
EEG karakteristike:
- između napada može biti normalno;
- usporavanje glavne pozadine;
- jednostrano suzbijanje alfa ritma uz bruto organske promjene;
- EEG obrasci se ne mijenjaju pri otvaranju očiju (diferencijalna dijagnoza sa idiopatskom okcipitalnom epilepsijom)
- širenje epiaktivnosti na vremenske vodove;
- provokacija - fotostimulacija.
Maligni migracijski žarišta ranog djetinjstva (Coppola sindrom - Dulac)
Relativno novi oblik fokalne epilepsije.
osobina:
- etiologija nepoznata (verovatno genetskog porekla);
- 6 godina starosti;
- normalan razvoj za debi;
- motorička i intelektualna regresija;
Napadi:
- fokalni motor;
- bilateralni tonik-klon;
- vegetativno (apneja, cijanoza)
- u obliku serija i klastera (2-5 dana), kratkih remisija.
progresivna mikrocefalija;
na EEG-u - tipičan fokalni uzorak u različitim vodovima;
MRI je normalna.
Lečenje: lekovi prve linije - topiramat, lamotrigin, drugi - valproična kiselina, levetiracetam.
Zaključci
Upotreba nove definicije epilepsije i nova klasifikacija napadaja omogućuju da se uzmu u obzir većina vrsta epileptičkih napadaja i da se termin "epilepsija" uskladi s terminologijom koju koristi većina liječnika koji se bave epilepsijom.
Posljednjih godina sintetizirano je mnogo novih antiepileptičkih lijekova kako bi se poboljšao kvalitet skrbi o pacijentima: u razdoblju od 2007. do 2012. godine. - ako je karbarbazepin acetat (eslikarbazepin acetat), lakosamid (lakozamid), perampanel (perampanel), retigabin (retigabin), rufinamid (rufinamid), stiripentol (stiripentol), 2016. - brivaracetam (brivaracetam). Ali u periodu djetinjstva antiepileptici sa širokim spektrom djelovanja ostaju zlatni standard - valproična kiselina, lamotrigin, topiramat i karbamazepin, koji su osnovni.
Epileptički sindromi kod djece: dijagnostika i liječenje
Mukhin Konstantin Y.
Petruhin Andrey Sergeevich
Epilepsija- jedan od najhitnijih problema u dječjoj neurologiji. Učestalost epilepsije u dječjoj populaciji je 0,5-0,75% dječje populacije, a febrilne konvulzije do 5%. Nažalost, treba napomenuti da u našoj zemlji do sada postoji velika konfuzija u uspostavljanju dijagnoze epilepsije, što značajno iskrivljuje statistiku. Dijagnoze kao što su "epileptiformni sindrom", "konvulzivni sindrom", "povećana konvulzivna spremnost", itd., Su, u stvari, epilepsija i treba da budu jasno sistematizovane u skladu sa Međunarodnom klasifikacijom. Svrha ove publikacije je da upozna lekare sa klasifikacijom, principima dijagnoze i lečenja glavnih oblika epilepsije kod dece.
Definicija Epilepsija je hronična moždana bolest koju karakterišu ponovljeni neprovocirani napadi motornih, senzornih, autonomnih, mentalnih ili mentalnih poremećaja koji su rezultat prekomernih neuralnih pražnjenja. Predstavljena definicija sadrži dva važna pitanja. Prvo, epilepsija ne uključuje pojedinačne napade, bez obzira na njihove kliničke manifestacije. Samo ponovljeni napadi su osnova za uspostavljanje dijagnoze epilepsije. Drugo, epilepsija uključuje spontane, neprovocirane napadaje (sa izuzetkom oblika refleksa). Po definiciji, febrilne konvulzije, kao i napadaji koji se javljaju kod akutnih bolesti mozga (na primjer, encefalitis, subduralni hematom, akutna cerebrovaskularna nesreća, itd.) Nisu epilepsija.
Klasifikacija. Manifestacije epilepsije su izuzetno raznovrsne, što je od samog početka proučavanja bolesti otežavalo stvaranje jedinstvene klasifikacije. Savremenu klasifikaciju epileptičkih napadaja usvojila je Međunarodna liga protiv epilepsije 1981. godine u gradu Kyoto (Japan). Za razliku od prethodnih klasifikacija, on uzima u obzir i kliničke i neurofiziološke (EEG) kriterije za većinu tipova epileptičkih napadaja (Tabela 1). Klasifikacija dijeli sve tipove epileptičkih napadaja na parcijalne (fokalne, fokalne, lokalne, lokalizirane), generalizirane i neklasifikovane. Parcijalni napadaji se dijagnosticiraju u slučaju kada na početku paroksizma postoje jasni klinički i elektrofiziološki kriteriji za uključivanje određenih moždanih struktura. U slučaju da napad počne kao parcijalni, a onda su zahvaćeni čitavi mišići trupa i ekstremiteta i znakovi uključenosti obje hemisfere na EEG, treba ga klasificirati kao fokalne sa sekundarnom generalizacijom.
Klasifikacija je pojasnila koncept jednostavnih i kompleksnih parcijalnih napada. Jednostavni parcijalni - napadi bez isključivanja svesti. Pod kompleksnim parcijalnim napadima treba shvatiti paroksizme sa potpunim gašenjem svijesti. Prema savremenoj klasifikaciji, svi napadi koji se javljaju kod fenomena depersonalizacije, stanja snova, kognitivnih poremećaja itd. Nisu jednostavni, već su delimični, jer se pacijentova svijest tokom tih paroksizama mijenja, ali se ne isključuje, a pamćenje napada se čuva. Takođe, klasifikacija iz 1981. godine predviđa da jedan pacijent može imati nekoliko
različite vrste napadaja. Naprimjer, napad, počevši od jednostavnog djelomičnog, može se transformirati u složeni parcijalni, a zatim u sekundarni-generalizirani. Iz klasifikacije je uklonjen izraz „polimorfni napadi“, koji ne sadrži nikakve informacije i nije preporučljiv za upotrebu. Prema tome, klasifikacija Kyota je u ovoj fazi najkompletnija sistematizacija epileptičkih napadaja.
Sa akumulacijom kliničkog iskustva, uvođenjem metode video-EEG monitoringa, razvojem neuro-snimanja, molekularne genetike i drugih znanosti u praksi, postalo je očigledno da postoji određen broj specifičnih oblika epilepsije koji imaju svoje kliničke karakteristike (tipične tipove napada), tok i prognozu. . Neki od ovih oblika su poznati već dugo vremena, kao što je Westov sindrom, Lennox-Gastautov sindrom, rolandička epilepsija. Drugi, benigni, porodični neonatalni napadi, teška infantilna mioklonična epilepsija, itd., Identifikovani su samo poslednjih godina. Ovi oblici epilepsije, ili, prema međunarodnoj klasifikaciji, epileptički sindromi, po pravilu se ne manifestiraju ni u jednoj vrsti napada, već po njihovoj kombinaciji. Epileptički sindromi su definisani kao odvojeni, nezavisni oblici epilepsije koje karakteriše ograničena starost napadaja, prisustvo posebnog tipa napadaja, specifične promjene u EEG-u (karakteristične za ovaj sindrom), obrasce tijeka i prognozu. Na primjer, jedan tip napadaja - absans - može biti uključen u strukturu niza epileptičkih sindroma: djeca i mladi su odsutni epilepsija, juvenilna mioklonična epilepsija, epilepsija s miokloničkim absansom, i drugi, a obilježja tijeka i prognoza za sve ove sindrome su različita.
Temeljno novi korak u razvoju epileptologije bio je stvaranje moderne klasifikacije "epilepsije, epileptičkih sindroma i povezanih sa napadima bolesti". Ova klasifikacija je usvojena od strane Međunarodne lige protiv epilepsije u oktobru 1989. godine u Nju Delhiju i sada je opšte prihvaćena za epileptologe širom sveta (Tabela 2).
Klasifikacija epileptičkih sindroma zasnovana je na sljedećim principima:
Princip lokalizacije:
lokalizacijski (fokalni, lokalni, parcijalni) oblici epilepsije;
generalizovani oblici;
forme koje imaju karakteristike i parcijalne i generalizovane.
Princip etiologije:
simptomatski,
kriptogeni,
idiopatski.
Starost napada:
oblici novorođenčadi,
dijete,
djeca,
mladenački
Glavni tip napadaja koji definira kliničku sliku sindroma:
apscesi
mioklonska apsantija,
infantilni grčevi, itd.
Značajke protoka i prognoze:
benigni;
teška (maligna).
Klasifikacija dijeli sve oblike epilepsije na simptomatske, idiopatske i kriptogene. Simptomatski oblici uključuju epileptičke sindrome sa poznatom etiologijom i potvrđene morfološke poremećaje (tumori, ožiljci, glioze, ciste, disgeneza, itd.). U idiopatskim oblicima nema bolesti koje mogu biti uzrok epilepsije, a epilepsija je nezavisna bolest. Trenutno utvrđeni genetski determinizam idiopatski oblici epilepsije. Termin "kriptogeni" (skriven) odnosi se na one sindrome, čiji uzrok ostaje skriven, nejasan. Ovi sindromi ne zadovoljavaju kriterijume za idiopatske oblike, ali nema dokaza o njihovoj simptomatskoj prirodi. Na primjer, u slučaju kombinacije epilepsije s hemiparezom ili oligofrenijom, pretpostavlja se simptomatska priroda bolesti, ali CT-om i MR-pregledom promjene u mozgu se ne vizualiziraju. Ovaj slučaj je klasifikovan kao kriptogeni. Očigledno, sa poboljšanjem tehničkih mogućnosti neuro-snimanja (npr. PET), većina kriptogenih oblika će biti prebačena u simptomatsku kategoriju.
Simptomi Semiologija i klinički prikaz različitih epileptičkih napada opisani su detaljno u mnogim priručnicima za epilepsiju. Međutim, u literaturi nije posvećena dovoljna pažnja dijagnozi pojedinih oblika epilepsije (epileptičkih sindroma). Osvrnimo se na kriterije dijagnoze i principe liječenja glavnih epileptičkih sindroma djetinjstva i adolescencije.
Pedijatrijska epilepsija apscesa
Absansy - tip generaliziranih ne-konvulzivnih napada, koji se odlikuju visokom učestalošću i kratkim trajanjem paroksizama sa nesvjesnošću i prisustvom specifičnog obrasca na EEG-u - generaliziranoj aktivnosti vršnog vala sa frekvencijom od 3 Hz.
Debi odsustva u apsantiji dječje epilepsije (AED) uočen je u rasponu starosti od 2 do 9 godina, u prosjeku 5,3 godine. Starosni vrhunac manifestacije je 4-6 godina, sa dominacijom djevojčica po spolu.
Klinički, izostanci se odlikuju iznenadnim kratkim isključenjem (ili značajnim smanjenjem nivoa) svijesti bez ili sa minimalnim motoričkim pojavama. Aura, kao i konfuzija nakon napada, nisu karakteristične. Trajanje odsustva varira od 2-3 do 30 sekundi, u prosjeku 5-15 sekundi. Karakteristična osobina apsensova je njihova visoka učestalost, dostiže desetine i stotine napada dnevno.
Princip je podjela odsutnosti na jednostavne i složene. Jednostavne apscese karakteriše prestanak svih aktivnosti, „smrzavanje“, „blijedi“ pacijenti, fiksni „odsutni“ izgled i zbunjeni hipomimetički izraz lica. Jednostavni apsani u klinička slika DAE je samo 20%. Za DAE kompleksnije kompleksne odsutnosti koje se javljaju sa minimalnom komponentom motora. Izdvajaju se sljedeći tipovi kompleksnih izostanaka: mioklonska, tonička, atonska, vegetativna komponenta, kao i automatizmi i fokalne pojave. Najčešće se navode izostanci sa miokloničnom i toničnom komponentom.
Odsustvo sa miokloničnom komponentom utvrđeno je kod 40% pacijenata sa AED. Manifest: mioklonija vek; perioralni mioklonus (ritmičko istezanje usana kao zlatna ribica); perinazalni mioklonus (ritmičko trzanje krila nosa). Kod velikog broja pacijenata tokom napada primijećeno je jednokratno trzanje ili nekoliko kratkih slabih trzanja mišića ramenog pojasa i / ili ruku.
Odsustva sa toničnom komponentom su najtipičniji za AED, a uočavaju se kod 50% pacijenata. Pojavljuje se odstupanjem glave, a ponekad i tijela, leđa (retro-pulsirajuće odsutnosti), tonske abdukcije očne jabučice gore ili u stranu. Ponekad se tokom napada javlja blaga tonička napetost (obično asimetrična) mišića gornjih ekstremiteta.
Absancije sa atonskom komponentom nisu karakteristične za AED i navode se samo u izolovanim slučajevima, uglavnom u atipičnim oblicima. Oni pokazuju nagli gubitak mišićnog tonusa u mišićima ruku (gubitak predmeta), vratu (pasivni kim), nogama (atono-astatski napadi). Atonska apsancija se češće naziva atipičnim apsanima (učestalost kompleksa vršnih talasa manjih od 2,5 Hz) u okviru Lennox-Gastaut sindroma.
Abscesi sa vegetativnom komponentom u prosjeku su uočeni u 5% bolesnika s AED. Manifestirana urinarna inkontinencija u vrijeme napada, midriaza, diskoloracija kože lica i vrata s pojavom urtikarijalnog osipa na koži ovih područja.
Odsustva sa fokalnom komponentom utvrđena su kod 15% pacijenata i značajno prevladavaju kod pacijenata sa toničnim absansom. Prilikom napada javlja se lagana jednostrana napetost mišića ruke ili lica, ponekad i pojedinačno myoclonic twitching; okretanjem glave i očiju u stranu. Treba imati na umu da je pojava izraženih fokalnih fenomena tokom napada alarmantna s obzirom na prisustvo parcijalnih paroksizama kod pacijenata. Automatizmi u strukturi izostanaka zapaženi su kod 35% pacijenata koji pate od AED. Najčešće se javljaju gestovi automatizma, faringo-oralnog i govornog.
Status odsutnosti u AED je zabilježen sa učestalošću od oko 10%. Ovo stanje se manifestuje oštrim povećanjem izostanaka, koji slijede jedan za drugim direktno ili u vrlo kratkom intervalu. Tu je amymia, drooling, motorna inhibicija (stupor). Trajanje statusa se kreće od nekoliko sati do nekoliko dana.
Generalizirani konvulzivni napadi (GSP) su nađeni u 1/3 pacijenata sa AED. Potrebno je nekoliko mjeseci ili godina od trenutka debi odsutnosti do pristupanja GSP-u. U većini slučajeva SHG-i se pridružuju 1-3 godine nakon početka bolesti (65% u grupi pacijenata sa SHGs), rjeđe u rasponu od 4-13 godina (35%). Preovladavaju retki, generalizovani toničko-klonični konvulzivni paroksizmi.
Čimbenici koji provociraju povećanje izostanaka uključuju sljedeće: hiperventilacija; lišavanje sna; fotostimulacija; intenzivna mentalna aktivnost ili, obrnuto, opušteno, pasivno stanje. Hiperventilacija - glavni faktor koji izaziva pojavu izostanaka. Provođenje 3-minutne hiperventilacije kod neliječenih pacijenata AED uzrokuje pojavu odsutnosti u gotovo 100% slučajeva; i kod pacijenata koji primaju AED, jedan je od kriterijuma za efikasnost terapije lekovima.
Učestalost detektovane epileptiformne aktivnosti u interiktalnom periodu sa AED je visoka i iznosi 75-85%. Najtipičniji EEG uzorak su bljeskovi generalizirane aktivnosti vrha. Učestalost kompleksa vršnih talasa varira od 2,5 do 4-5 u sekundi. (obično 3 Hz - tipični absans).
Tretman. Potpuna terapijska remisija kod AED-a se postiže u 70-80% slučajeva, a kod preostalih pacijenata zapaženo je značajno smanjenje napadaja. Liječenje uvijek treba započeti valproičnom kiselinom. Prosječna doza je 30-50 mg / kg / dan. u 3 doze. Takođe je prikazana visoka efikasnost suximida u zaustavljanju izostanaka. Prosječna doza lijeka je 15 mg / kg / dan. u 2-3 doze. Značajna negativna tačka u terapiji sa suksimidom je potpuno odsustvo dejstva leka na SHG. Kada su izostanci otporni na monoterapiju valproatima i sukcinimidima, propisana je kombinacija valproata i suksimida, valproata i lamotrigina (Lamictal). Prosečna dnevna doza Lamictala u kombinaciji sa valproatom je 1-5 mg / kg / dan. u 2 doze. Upotreba karbamazepina (finlepsin, tegretol, timonil) je strogo kontraindicirana u svim oblicima odsutne epilepsije zbog velike vjerovatnoće povećanja napadaja.
Epilepsija juvenilnog apscesa
Juvenilna apsantija epilepsija (UAE) je tip idiopatske generalizovane epilepsije, karakterizirana glavnim tipom napadaja - odsustvo debitiranja u pubertetskom periodu s velikom vjerovatnoćom spajanja SHG i karakterističnim EEG promjenama u obliku generalizirane vršne aktivnosti s frekvencijom od 3 Hz ili više.
Debitacija odsustva u SAE varira od 9 do 21 godine, u prosjeku 12,5 godina. Kod velike većine pacijenata (75%), izostanci počinju u relativno kratkom vremenskom intervalu - 9-13 godina. Važna karakteristika YuAZ-a je česti debut bolesti sa SHG-ima - 40% slučajeva.
Absansija kod pacijenata koji pate od SAE, manifestuje se kratkim prekidom svijesti sa kongelacijom i hipomimijom. Karakteristično je prevlast jednostavnih izostanaka, tj. Napadaji bez motorne komponente. Trajanje napada se kreće od 2 do 30 sekundi, u proseku 5-7 sekundi. Međutim, polovina pacijenata je imala vrlo kratke apsane, ne prelazeći 3 sekunde. Karakteristična karakteristika UAE je relativno niska učestalost napada u odnosu na AED. Kod većine pacijenata, tokom dana preovladavaju pojedinačni apscesi ili jedan napad nakon 2-3 dana.
Generalizirani konvulzivni napadaji su utvrđeni kod većine pacijenata - 75%. U grupi bolesnika sa SHG-om bolest se češće pojavljuje ne sa odsustvom, već sa tonično-kloničkim konvulzivnim paroksizmima. SHG karakterišu kratke, retke toničko-klonične konvulzije, koje se obično javljaju prilikom buđenja ili zaspanja.
Za razliku od DAE, hiperventilacija izaziva pojavu odsustva u ne više od 10% bolesnika sa SAE. GSP kod 20% pacijenata je izazvan deprivacijom sna.
Kada je ZZG studija u interiktalnom periodu, rezultati su blizu normalnog, navodi se u 25% pacijenata. Glavni EEG uzorak je generalizirana aktivnost vršnih valova sa frekvencijom od 3 Hz ili više (4-5 u sekundi), koja je uglavnom simetrična i bilateralno-sinhrona u prirodi.
Tretman. Efikasnost tretmana sa UAE je značajno niža nego kod DAE. Terapijska remisija se postiže u prosjeku kod 60% pacijenata, značajno smanjenje napadaja - 35%, bez učinka - 5%. Lečenje počinje monoterapijom valproičnom kiselinom. Prosječna doza je 30-50 mg / kg / dan. Zbog ekstremno velike vjerovatnoće pristupanja SHG-a u slučaju UAE, nije naznačeno da se započne liječenje sa sukcinimidima, kao i da se primijene kao monoterapija. U odsustvu značajnog efekta monoterapije valproatima u dovoljno visokim dozama, koristi se kombinacija valproata sa sukcinimidima ili lamictalom. Prosječna doza suksimide je 20 mg / kg / dan; Lamictal - 1-5 mg / kg / dan.
Epilepsija sa izolovanim generaliziranim konvulzivnim napadima
Epilepsija sa izolovanim SHG-om je definisana kao sindrom idiopatske generalizovane epilepsije, koja se manifestuje jednim tipom napadaja - primarno generalizovanim toničko-kloničkim konvulzivnim paroksizmom u odsustvu aure i jasnim fokusom na EEG.
Debi bolesti se posmatra u veoma širokom rasponu godina: od 1 do 30 godina sa maksimumom u pubertalnom periodu (prosječno -13,5 godina).
Klinički, GSP se manifestuje iznenadnim (bez aure) gašenjem svijesti sa padom pacijenata, konvulzijama, uspostavljanjem očnih jabučica, proširenih zjenica. Prvo dolazi kratka tonička faza, koja se pretvara u dužu kloničku fazu, nakon čega slijedi zapanjujući napad. Trajanje GSP-a je od 30 sekundi do 10 minuta (u prosjeku 3 minute). Učestalost napada je niska - od pojedinačne godišnje do 1 puta mjesečno, bez tendencije ka serijskoj i statusnoj struji. Karakterizira se vremenskim rasporedom većine napada do perioda buđenja i, retko, spavanja. Najznačajniji precipitacijski faktor je deprivacija sna i naglo nasilno buđenje. Možda češći napadaji u perimenstrualnom periodu.
EEG studija u interiktalnom periodu kod polovine pacijenata (!) Može biti unutar normalnih granica. Karakterizira ga generalizirana aktivnost vršnih valova s frekvencijom od 3 Hz i više, često s asimetrijom amplitude ili bifrontalnom dominacijom. Mogu se identifikovati različiti regionalni obrasci.
Tretman. Remisija se postiže kod 75-80% pacijenata. Osnovni lekovi su karbamazepin i valproati. U odsustvu generalizovane aktivnosti vršnog talasa na EEG, tretman počinje karbamazepinom, koji je efikasniji od valproata. Pri spajanju generaliziranih konvulzivnih napadaja izostanaka ili mioklonskih paroksizama, ili kada se na EEG-u pojavi generalizirana aktivnost vršnih valova, osnovni preparat je samo valproat. Prosečna doza karbamazepina je 15-25 mg / kg / dan. u 3 doze; Valproat 20-50 mg / kg / dan. u 3 doze. Rezervni lijekovi uključuju barbiturate (fenobarbital 1,5-3,0 mg / kg / dan u 1-2 doze; heksamidin, benzon), hidantoine (difenin 4-8 mg / kg / dan u 2 doze). U rijetkim rezistentnim slučajevima moguće su kombinacije: karbamazepin + valproati; karbamazepin + lamotrigin, karbamazepin + barbiturati; valproati + barbiturati. Uz neadekvatno lečenje, moguće je pridržavanje GSP izostanaka ili mioklonskih napada sa transformacijom u juvenilnu miokloničnu epilepsiju.
Juvenilna mioklonična epilepsija
Juvenilna mioklonična epilepsija (UME) je jedan od oblika idiopatske generalizovane epilepsije, karakteriziran debutom u adolescenciji sa pojavom masivnih bilateralnih miokloničnih napadaja, uglavnom u rukama, u periodu nakon buđenja pacijenata. YuME je jedan od prvih oblika epilepsije sa poznatim genetskim defektom. Pretpostavljen je model nasleđivanja sa dva lokusa (dominantno-recesivan), sa dominantnim genom koji se nalazi na kratkom kraku hromozoma 6.
Debitni UME varira od 7 do 21 godine sa maksimumom u uzrastu od 11-15 godina. Bolest može početi u ranijoj dobi od odsutnosti ili SHG-a, nakon čega slijedi dodavanje miokloničnih napadaja tijekom puberteta. Mioklonične napade karakterišu grmljavinska trzanja različitih mišićnih grupa; oni su češće bilateralni, simetrični, pojedinačni ili višestruki, varirajući u amplitudi. Lokalizira se uglavnom u ramenom pojasu i rukama, uglavnom u mišićnim grupama ekstenzora. Tokom napada, pacijenti bacaju predmete iz ruku ili ih bacaju u stranu. Kod 40% pacijenata mioklonični napadaji zahvaćaju mišiće nogu, dok se pacijent osjeća kao iznenadni udarac ispod koljena i lagano čučnjevi ili padovi (mioklono-astatski napadi); onda se odmah diže. Svest tokom napada obično se čuva. Mioklonični napadi se javljaju ili postaju ujutro, nakon što se pacijent probudi. U 90% slučajeva, oni su kombinovani sa GSP buđenjem, au 40% sa apsanima. Glavni faktori koji izazivaju napade su deprivacija sna i naglo nasilno buđenje. Približno 1/3 pacijenata sa UME (češće žene) pokazuju fotosenzitivnost.
Epileptiformna aktivnost na EEG detektovana je u 85% bolesnika u interiktalnom periodu. Najtipičnija je generalizovana brza (od 4 Hz i više) polipična valna aktivnost u obliku kratkih bljeska. Moguća je i pojava aktivnosti vršnog vala od 3 Hz.
Tretman. Zajedno sa terapija lekovima neophodno je strogo se pridržavati poštovanja sna i budnosti; izbegavajte lišavanje sna i faktore fotostimulacije u svakodnevnom životu. Osnovni lekovi su isključivo derivati valproične kiseline. Prosečna dnevna doza je -40-60 mg / kg.U slučaju nedostatka efikasnosti, propisana je polieterapija: valproat + suxilep (sa rezistentnim apsanima); valproat + fenobarbital ili heksamidin (za rezistentne SHG); valproroat + lamotrigin ili klonazepam (za rezistentne mioklonične napadaje i izraženu fotosenzitivnost).
Kompletna remisija leka se postiže kod 75% pacijenata, au većini slučajeva sa monoterapijom valproatom. Međutim, kasnije, sa ukidanjem AEP-a, recidivi su navedeni u polovini pacijenata. Kod ovog oblika epilepsije, preporučuje se da se AEP poništi nakon najmanje 4 godine od trenutka remisije.
Benigna djelomična epilepsija kod djece sa središnjim temporalnim vrhovima (rolandska epilepsija).
Rolandic epilepsija (ER) je idiopatska parcijalna epilepsija djetinjstva, koju karakteriziraju uglavnom kratki hemifacijalni motorički noćni napadi, često s prethodnom somatosenzornom aurom i tipičnim EEG promjenama.
RZ debi varira u rasponu od 2 do 14 godina. U 85% slučajeva napadaji počinju u dobi od 4-10 godina, a maksimalno 9 godina.
Posmatrani su jednostavni parcijalni (motorički, senzorni, autonomni), kompleksni parcijalni (motorički) i sekundarno-generalizirani konvulzivni napadaji. Najčešći su jednostavni parcijalni motorni i / ili senzorni paroksizmi. Početak napada iz somatosenzorne aure je karakterističan: peckanje, obamrlost na jednoj strani ždrela, jezik, desni. Zatim se pojavljuju motoričke pojave: unilateralni tonički, klonički ili toničko-klonički konvulzije mišića lica, usana, jezika, ždrijela, larinksa; pharingo-oral napada, često u kombinaciji s anarthria i hipersalivacija. U isto vrijeme, u snu, pacijenti emitiraju osebujna grla zvuči kao "grgljanje", "grunting", "gargling". Kod 20% pacijenata, konvulzije se mogu proširiti iz mišića lica u homolateralnu ruku (brahiofacijalni napadi) iu oko 8% slučajeva uključiti nogu. Kako bolest napreduje, napadaji mogu promijeniti stranu. Sekundarni generalizirani konvulzivni napadaji su uočeni u 20-25% bolesnika s OM. Trajanje napada sa re je malo: od nekoliko sekundi do 2-3 minuta. Učestalost je obično niska - u prosjeku 2-4 puta godišnje. U prvim mjesecima nakon početka bolesti napadaji mogu biti češći, ali s vremenom se pojavljuju sve manje i manje, čak i bez liječenja. Paroksizmi ER su "rigidno" povezani sa ritmom sna i budnosti. Najtipičniji noćni napadi, koji se javljaju uglavnom pri spavanju i buđenju. Samo u 15-20% pacijenata, konvulzije se primećuju iu snu iu budnom stanju.
Na EEG-u, u interiktalnom periodu, detektuju se karakteristične "rolandske" komplekse vrhunaca sa visokom frekvencijom, pri čemu je neophodno očuvati osnovnu aktivnost. Ovi kompleksi su spori, difuzni pikovi visoke amplitude ili oštri valovi (150–300 μV), često s kasnijim sporim valovima, s ukupnim trajanjem od oko 30 ms. Ovi kompleksi nalikuju zubima ORS EKG. Rolandic kompleksi se obično nalaze u centralnim i temporalnim regionima; može se posmatrati i unilateralno (obično kontra-lateralno do hemifacijalnih napadaja) i bilateralno nezavisno. Tipična je nestabilnost EEG uzoraka, njihova varijabilnost od jednog do drugog zapisa.
Tretman. Osnovni lek je valproat. Prosječna doza je 20-40 mg / kg / dan. u 3 doze. U slučaju neefikasnosti - prelazak na karbamazepin-10-20 mg / kg / dan. u 2-3 doze. Politerapija nije dozvoljena!
Potpuna terapijska remisija se postiže u skoro 100% slučajeva. Nakon 14 godina, napadi nestaju (sa terapijom ili spontano) kod 93% pacijenata, a nakon 16 godina - kod 98% pacijenata. S obzirom na apsolutno povoljnu prognozu, neki autori predlažu da se ne propisuje lečenje za ustanovljenu dijagnozu ER. Ovo gledište je sporno.
Idiopatska parcijalna epilepsija sa zatiljnim paroksizmima
Idiopatska parcijalna epilepsija sa okcipitalnim paroksizmom (benigna okcipitalna epilepsija (DZE)) je oblik idiopatske lokalizacije vezane za dječju epilepsiju, koju karakteriziraju jednostavni parcijalni napadaji s oštećenjem vida i prisutnost specifične aktivnosti vrha valova u okcipitalnoj spazmodičnoj epilepsiji.
Bolest počinje u dobi od 2-12 godina sa dva debi debla - oko 3 i 9 godina. Tipični parcijalni senzorni paroksizmi sa vidnim poremećajima su tipični. Karakteristične su jednostavne vizuelne halucinacije, fotopsije, vizuelne iluzije (makro, mikropsija). Moguća je pojava prolazne amauroze i istoimene kvadrantne hemiopatije. Tokom napada, višestruka komponenta se često posmatra sa nasilnim okretom očiju i glave. Kada se lezija u potiljačnom režnju često posmatra iradijacija ekscitacije anteriorno (u temporalnom i frontalnom režnju) sa pojavom složenih strukturnih halucinacija; deaktivacija svijesti i pojava sekundarnih generaliziranih konvulzivnih napadaja. Tipična pojava napadaja u snu, posebno kod buđenja pacijenata. Napadi često prate simptome migrene: glavobolja i povraćanje. Prvi napadi kod male djece su najteži i produženi (do nekoliko sati pa čak i dana).
EEG se određuje pojavom visokih amplitudnih aktivnosti vršnih talasa u jednom od okcipitalnih tragova, ili bioccipitacijom nezavisno. Morfologija uzoraka nalikuje na rolandsku epilepsiju. Karakterizira se nestankom epiaktivnosti prilikom snimanja EZG-a sa otvorenim očima.
Tretman. Lijekovi izbora su derivati karbamazepina. Prosječna dnevna doza je oko 20 mg / kg. Kod neefikasnosti se koristi valproat - 30-50 mg / kg / dan. Rezervni lijek fenitoin - 3-7 mg / kg / dan. Lečenje se vrši samo monoterapijom. Rezistentni slučajevi moraju se razlikovati od simptomatske okcipitalne epilepsije. Potpuna terapijska remisija je uočena u 95% slučajeva. Istovremeno, napadi se zaustavljaju, a doza AEP je veća nego kod rolandske epilepsije.
Westov sindrom
Westov sindrom (SV) je rezistentni oblik generalizovane epilepsije u ranom djetinjstvu, karakteriziran napadima u obliku infantilnih spazama, odgođenim psihomotornim razvojem i specifičnim promjenama ZZG (hypsrythmia). Starosni vrhunac manifestacije - 4-7 mjeseci. Razlikuju se simptomi (malformacije mozga, tubularna skleroza, perinatalna encefalopatija, tezaurizam, itd.) I kriptogeni oblici. U simptomatskom obliku, psihomotorni razvoj djece obično pati od rođenja, s kriptogenim - od trenutka početka napada.
Infantilni grčevi se manifestuju naglim kontrakcijama mišića vrata, trupa, udova, koji su obično bilateralni i simetrični. Flexor grčevi ("Salaam napadi") sa savijanjem vrata, trupa, ruku su najtipičniji; fleksija, adukcija i podizanje nogu. Napadi su kratki, grupisani u seriju; češće se javljaju odmah nakon buđenja pacijenata.
U neurološkom statusu otkriveno je veliko odlaganje mentalnog i motoričkog razvoja, često tetrapiramidna simptomi.
Na EEG-u je zabeležena gypsarythmia - visoko-amplitudna nepravilna, slabo sinhronizovana aritmička spora vala sa šiljcima.
Tretman Počinje sa Valproatom, Vigabatrinom (Sabril) ili ACTH. U slučajevima kriptogenog SV, ACTH (ili Sinakten Depot) se primjenjuje u dozi od 0,1 mg / kg / dan. ili prednizon - 2-5 mg / kg / dan. U simptomatskim CB-valproatima (50-100 mg / kg / dan i više) kao monoterapija ili u kombinaciji sa ACTH. Poslednjih godina, prema mnogim autorima, preporučuje se upotreba vigabatrina u dozi od 100 mg / kg / dan. Vigabatrin je lijek apsolutnog izbora u liječenju SV, uzrokovan tubularnom sklerozom. U odsustvu efekta monoterapije, bazični AED se kombinuju sa lamotriginom, karbamazepinom ili benzodiazepinima.
Prognoza bolesti je ozbiljna, kako u smislu napadaja tako i psihomotornog razvoja. Većina djece je invalid, nije sposoban za samostalan život. S vremenom se SV transformiše u SLH (1/3 slučajeva) ili multifokalnu epilepsiju.
Lennox-Gasto sindrom
Lennox-Gasta sindrom (SLH) je epileptička encefalopatija djetinjstva, koju karakterizira polimorfizam napadaja, kognitivno oštećenje, specifične EEG promjene i otpornost na terapiju. Incidencija SLH je oko 5% među svim oblicima epilepsije u djece i adolescenata; Dečaci su češće bolesni.
Bolest se dešava uglavnom u dobi od 2-8 godina (obično 4-6 godina). Ako se SLH razvije tokom transformacije sa Westovog sindroma, onda postoje 2 moguće opcije:
1. Infantilni grčevi se pretvaraju u tonične napade u odsustvu latentnog perioda i glatko prelaze u SLG.
2. Infantilni grčevi nestaju; psihomotorni razvoj djeteta je donekle poboljšan; EEG slika se postepeno normalizuje. Zatim dolazi latentni period koji varira u trajanju kod različitih pacijenata; postoje napadi iznenadnih kapi, atipične izostanke, i raste difuzna aktivnost pik-valova na GWD-u.
Za SLG karakteristična je triada napada: paroksizmi padova (atonični i mio-klonički-astatički); tonički napadi i abnormalne izostanke. Najčešći napadi iznenadnog pada usled toničkih, miokloničkih ili atonskih (negativnih mioklonusa) paroksizama. Svest se može sačuvati ili nakratko isključiti. Nakon pada, ne primećuju se grčevi, a dijete se odmah diže. Učestali padovi vode do teških trauma i invalidnosti pacijenata.
Tonični napadaji su aksijalni, proksimalni ili totalni; simetrični ili jasno lateralizirani. Napadi uključuju naglu fleksiju vrata i trupa, podizanje ruku u stanju polu-infleksije ili produžetka, produžetak nogu, kontrakciju mišića lica, rotacione pokrete očnih jabučica, apneju, crvenilo lica. Mogu se pojaviti tokom dana, a naročito često noću.
Atipična apsancija je takođe bila karakteristična za SLG. Njihove manifestacije su različite. Svijest je nepotpuna. Neki stepen motoričke i govorne aktivnosti može da traje. Postoji hipomimija, balavljenje; stoljeće myoclonus, usta; atoni fenomeni (glava pada na grudi, usta se razdvajaju). Atipična apsancija je obično praćena smanjenjem mišićnog tonusa, koji uzrokuje neku vrstu “slabljenja” tela, počevši od mišića lica i vrata.
U neurološkom statusu su uočene manifestacije piramidalne insuficijencije, poremećaji koordinacije. Karakterizira se smanjenjem inteligencije, ali ne dosežu, međutim, teške. Intelektualni nedostatak utvrđuje se od rane dobi, pre bolesti (simptomatski oblici) ili se razvija odmah nakon početka napada (kriptogeni oblici).
Kod EEG studije u velikom procentu slučajeva otkrivena je nepravilna difuzna, često sa amplitudnom asimetrijom, spora aktivnost vrha sa frekvencijom od 1,5-2,5 Hz tokom budnosti i brza ritmička pražnjenja sa frekvencijom od oko 10 Hz tokom sna.
U neuro-slikovnom prikazu mogu se javiti različite strukturne abnormalnosti u moždanoj kori, uključujući malformacije: hipoplazija corpus callosum, hemimegalenephalus, kortikalna displazija itd.
U tretmanu SLH treba da izbegava kognitivno-supresivne lekove (barbiturate). Valproati, lamotrigin, karbamazepin, benzodiazepini se najčešće koriste za SLH. Tretman započinje derivatima valproične kiseline, postepeno ih povećavajući do maksimalne tolerantne doze (70-100 mg / kg / dan i više). Karbamazepin je efikasan za tonične napadaje - 15-30 mg / kg / dan, ali može povećati izostanke i mioklonične paroksizme. Jedan broj pacijenata reaguje na povećanje doze karbamazepina sa paradoksalnim povećanjem napadaja. Benzodiazepini imaju efekat na sve vrste napadaja, ali ovaj efekat je privremen. U grupi benzodiazepina koriste se klonazepam, klobazam (frisium) i nitrazepam (radedorm). Kod atipične apsanije, suxilep može biti efikasan (ali ne kao monoterapija). Pokazali smo visoku efikasnost kombinacije valproata sa lamictalom (1-5 mg / kg / dan i više) (Tabela 5). Ova terapija je optimalna kako u smislu efikasnosti tako i tolerancije (ne inhibira kognitivne funkcije). U SAD se široko koristi kombinacija valproata i felbamata (Talox).
Prognoza za SLH je teška. Kontinuirana konvulzivna kontrola se postiže samo u 5-15% pacijenata. Prognostički povoljna je prevalencija miokloničnih napadaja i odsustvo velikih strukturnih promjena u mozgu; negativni faktori su dominacija toničkih napada i bruto intelektualni deficit.
Simptomatska parcijalna epilepsija
U simptomatskoj parcijalnoj epilepsiji otkrivene su strukturne promjene u cerebralnom korteksu. Razlozi koji određuju njihov razvoj su raznovrsni i mogu se predstaviti sa dvije glavne grupe: perinatalnim i postnatalnim faktorima. Anamneza perinatalne encefalopatije nađena je kod 35% pacijenata; Postnatalni faktori uključuju neuroinfekciju, traumatsku povredu mozga i tumore mozga.
Početak napada sa simptomatskom parcijalnom epilepsijom varira u širokom rasponu godina, s maksimumom u predškolskom uzrastu. Ove slučajeve karakterišu promjene u neurološkom statusu, često u kombinaciji sa smanjenjem inteligencije; pojava regionalnih obrazaca na EEG, otpornost napada na AEP. Razlikuju se simptomatski parcijalni oblici epilepsije: temporalna, frontalna, parijetalna i okcipitalna. Prva dva su najčešća i čine 80% svih slučajeva.
Simptomatska epilepsija temporalnog režnja
Kliničke manifestacije VE su izuzetno različite. U nekim slučajevima, atipični febrilni napadi prethode razvoju bolesti. CE se manifestuje jednostavnim, kompleksnim parcijalnim, sekundarno-generaliziranim napadima ili njihovom kombinacijom. Posebno je karakteristično prisustvo kompleksnih parcijalnih napadaja koji se javljaju sa poremećajem svijesti, u kombinaciji s netaknutom, ali automatiziranom motoričkom aktivnošću. Automatizmi u kompleksnim parcijalnim napadajima mogu biti jednostrani, nastaju na homolateralnoj strani, a često se kombiniraju s distoničkim položajem ruke na kontralateralnoj strani. CE se dijele na amigdalo-hipokampalni (paleokortikalni) i lateralni (neokortikalni) epilepsiju.
Epilepsija amigdala-hipokampalnog temporalnog režnja karakterizirana je pojavom napadaja s izoliranim poremećajem svijesti. Došlo je do smrzavanja pacijenata sa licem nalik maski, široko otvorenih "zbunjenih" očiju i posmatrano u jednom trenutku. Istovremeno se mogu navesti različiti vegetativni fenomeni: blanširanje lica, širenje zjenice, znojenje, tahikardija. Postoje 3 tipa SPP sa izolovanim poremećajem svijesti:
isključivanje svesti sa smrzavanjem i iznenadnim prekidom motoričkih i mentalnih aktivnosti;
isključivanje svesti bez prekidanja motoričke aktivnosti;
isključivanje svesti sa sporim padom ("šepajući") bez napadaja ("temporalna sinkopa").
Značajni su i vegetativno-visceralni paroksizmi. Napadi se manifestuju nelagodnošću u trbuhu, bolovima u pupku ili epigastriumu, tutnjanjem u abdomenu, poticanjem stolice, ispuštanjem gasa (epigastrični napadi). Možda pojava "rastuće epileptičke senzacije", koju pacijenti opisuju kao bol, žgaravicu, mučninu, koji dolazi iz trbuha i podiže se u grlo, sa osećajem suženja, kompresijom vrata, grudanjem u grlu, često sa kasnijim isključenjem svesti i grčeva. Kada su uključeni u proces kompleksa u obliku badema, javljaju se napadi straha, panike ili besa; iritacija kuka uzrokuje olfaktorne halucinacije. Mogući su napadi sa poremećenim mentalnim funkcijama (stanja sanjanja, već viđena ili nikad viđena, itd.).
Lateralna VE se manifestuje napadima sa oštećenjem sluha, vida i govora. Karakteristična je pojava svetlo obojenih strukturnih (za razliku od okcipitalne epilepsije) vizuelnih halucinacija, kao i složenih auditivnih halucinacija. Oko 1/3 žena koje pate od VE, zabeležilo je povećanje napada u perimenstrualnom periodu.
Neurološki pregled djece oboljele od VE često otkriva mikrofokalne simptome, kontralateralni fokus: neispravnost 7. i 12. kranijalnih živaca centralnog tipa, revitalizacija refleksa tetiva, pojava patoloških refleksa, blagi fokalni poremećaji, itd. , manifestuje se pretežno intelektualno-mentički ili emocionalno-lični poremećaji; karakteriziran pojavom teških poremećaja pamćenja. Očuvanje inteligencije uglavnom zavisi od prirode strukturnih promena u mozgu.
U EEG studijama, vršni ili češće uporni regionalni sporo-talasni (theta) aktivnost je uočena u privremenim vodama, obično s prednjom distribucijom. Kod 70% pacijenata otkriveno je naglašeno usporavanje glavne aktivnosti pozadinskog snimanja. Kod većine pacijenata se vremenom epileptička aktivnost posmatra kao bitemporalna. Da bi se identifikovala lezija lokalizovana u medijsko-bazalnim područjima, poželjno je koristiti invazivne sfenoidne elektrode.
Neuroimaging otkriva razne makrostrukturne abnormalnosti u mozgu. Najčešći nalaz u MRI skeniji je medijska temporalna (incizuralna) skleroza. Često je takođe bila izražena lokalna ekspanzija brazdi, smanjenje volumena zahvaćenog temporalnog režnja, parcijalna ventrikulomegalija.
TretmanVE - težak zadatak; mnogi pacijenti su otporni na terapiju. Osnovni lekovi su derivati karbamazepina. Prosječna dnevna doza je 20 mg / kg. U slučaju neefikasnosti, povećati dozu na 30 mg / kg / dan. i više prije izgleda pozitivan efekat ili prvi znaci opijenosti. U odsustvu efekta, primenu karbamazepina treba prekinuti, umesto imenovanja fenitoina za kompleksne parcijalne napade ili valproata za sekundarne generalizirane paroksizme. Doziranje difenina u tretmanu VE je 8-15 mg / kg dnevno, valproat-50-100 mg / kg / dan. U odsustvu efekta monoterapije, moguća je upotreba polieterapije: karbamazepin + valproati, karbamazepin + lamictal, karbamazepin + fenobarbital, fenobarbital + difenin (potonja kombinacija je manje poželjna i uzrokuje značajno smanjenje pažnje i pamćenja, posebno kod djece). Pored osnovne terapije za AED kod žena, mogu se koristiti i spolni hormoni, posebno efikasni za menstrualnu epilepsiju. Oksprogesteron kapronat se koristi 12,5% rastvor od 1-2 ml intramuskularno jednom za 20-22 dana menstrualnog ciklusa.
Prognoza zavisi od prirode strukturnog oštećenja mozga. Sa godinama, većina pacijenata raste uporni mentalni poremećaji, značajno komplicirajući socijalnu adaptaciju. Generalno, oko 30% pacijenata koji pate od ET su otporni na tradicionalnu anti-epileptičnu terapiju i kandidati su za neurokiruršku intervenciju.
Simptomatska frontalna epilepsija
Klinički simptomi frontalne epilepsije (PE) su različiti. Bolest se manifestuje jednostavnim i kompleksnim parcijalnim napadima, kao i, što je posebno karakteristično, sekundarnim generaliziranim paroksizmima. Razlikuju se sledeći oblici LE: motorički, operularni, dorzolateralni, orbitofrontalni, prednji prednji polarni, cingularni, koji potiču iz dodatne motorne zone.
Motorni paroksizmi se javljaju tokom iritacije prednjeg centralnog girusa. Karakteristični napadi Jacksona, razvoj kontralateralnog nidusa. Konvulzije su pretežno klonske prirode i mogu se širiti u obliku uzlaznog (licem-za-lice) ili silaznim (licem-u-stopu) maršem; u nekim slučajevima sa sekundarnom generalizacijom. Sa lezijom u paracentralnim lobulama, mogu se uočiti konvulzije u ipsilateralnom ekstremitetu ili bilateralne. Slaba slabost u udovima (Toddova paraliza) je čest fenomen LE.
Operativni napadi javljaju se tokom stimulacije operularne zone donjeg frontalnog girusa na spoju sa temporalnim režnjem. Pojavljuje se paroksizmalno žvakanje, sisanje, pokreti gutanja, smacking, lizanje, kašljanje; karakteristična je hipersalivacija. Možda ipsilateralno trzanje mišića lica, oštećenje govora ili nevoljna vokalizacija.
Dorzolateralni napadaji se javljaju kod iritacije gornjeg i donjeg prednjeg mozga. One se manifestuju neželjenim napadima sa nasilnim okretanjem glave i očiju, obično kontra lateralna iritacija. Uz zahvaćanje stražnjih dijelova donjeg frontalnog girusa (Broka centar), uočeni su paroksizmi motorne afazije.
Orbitofrontalni napadi javljaju se tokom iritacije orbitalnog korteksa donjeg frontalnog gyrusa i manifestuju se nizom vegetativno-visceralnih fenomena. Karakterizira ih epigastrični, kardiovaskularni (bol u srcu, promena srčanog ritma, krvni pritisak), respiratorna (inspiratorna dispneja, osećaj gušenja, kompresija u vratu, "koma" u grlu). Često postoje faringo-oralni automatizmi sa hipersalivacijom. Obilje vegetativnih fenomena u strukturi napada privlači pažnju: hiperhidroza, bljedilo kože, često sa hiperemijom lica, oslabljena termoregulacija, itd. Moguće je pojavljivanje tipičnih kompleksnih parcijalnih (psihomotornih) paroksizama sa automatizmom pokreta.
Prednji (pred-polarni napadaji se javljaju kada je iritiran štit frontalnog režnja. Karakterišu ih jednostavni parcijalni napadaji sa oštećenim mentalnim funkcijama. Oni se manifestuju naglim "neuspehom misli", "prazninom u glavi", konfuzijom ili, naprotiv, nasilnim pamćenjem, bolnim, bolnim osećajem potrebe za nečim Snažan „priliv misli“, „vrtlog ideja“ je moguć - osećaj iznenadnog pojavljivanja u umu misli koje nisu u sadržaju povezane sa trenutnom mentalnom aktivnošću. Moguće je riješiti se tih misli prije kraja napada.
Cingularni napadi potiču iz prednjeg dijela cingularnog girusa medijalnog područja frontalnih režnjeva. Oni se manifestuju uglavnom u složenim, rjeđe u jednostavnim parcijalnim napadima sa kršenjem ponašanja i emocionalne sfere. Karakteriziraju se teški parcijalni napadi automatizmom gestova, crvenilom lica, izrazom straha, ipsilateralnim treptajućim pokretima, a ponekad i kloničkim konvulzijama kontralateralnih ekstremiteta. Možda pojava paroksizmalnih disforičnih epizoda sa gadnošću, agresivnošću, psihomotornom agitacijom.
Napade koji potiču iz dodatne motorne zone prvi su opisali Penfield, ali tek nedavno sistematizirani. Ovo je prilično česta vrsta napada, posebno kada uzmete u obzir paroksizme koji se javljaju u drugim dijelovima frontalnog režnja, često zračeći u dodatnu motornu zonu. Karakteristično je prisustvo čestih, obično noćnih, jednostavnih parcijalnih napada alternativnim hemikonvulzijama, arhaičnim pokretima; konvulzije sa prestankom govora, fuzzy, slabo lokalizovane senzorne senzacije u trupu i ekstremitetima. Parcijalni motorički napadi obično se manifestuju kao tonički konvulzije koje se javljaju s jedne ili druge strane, ili bilateralno (izgledaju kao generalizirane). Karakteristična tonička napetost sa usponom kontralateralne ruke, adverzija glave i očiju (pacijent gleda u njenu podignutu ruku, kakva jeste). Opisana je pojava "inhibitornih" napada sa paroksizmalnom hemiparezom. Napadi arhaičnih pokreta obično se javljaju noću sa visokom frekvencijom (do 3-10 puta po noći, često svake noći). Karakterišu ih iznenadno buđenje pacijenata, krik, grimasa užasa, motivska oluja: mahanje rukama i nogama, boks, pedaliranje (slično biciklizmu), pokreti karlice (kao u koitusu), itd. Ove napade treba razlikovati od histeričnih i paroksizmalnih noćnih strahova kod djece.
EEG studija sa PE može pokazati sljedeće rezultate: normalna aktivnost, vršna valna aktivnost ili usporavanje (periodično ritmično ili kontinuirano) regionalno u frontalnim, frontalno-centralnim ili frontalno-temporalnim vodovima; bifrontalni nezavisni žarišta vršnog vala; sekundarna bilateralna sinhronizacija; brza (beta) aktivnost regionalne frontalne niske amplitude. Lomovi lokalizirani u orbitofrontalnoj, operularnoj i pomoćnoj motornoj zoni ne smiju se mijenjati kada se primjenjuju površinske elektrode i zahtijevaju upotrebu dubinskih elektroda ili kortografije. Porazom dodatne motorne zone ZEG, obrasci su često ipsilateralni do napada ili bilateralnih, ili postoji fenomen sekundarne bilateralne sinhronizacije Jasper-a.
Tretman PE se sprovodi u skladu sa opštim principima lečenja epilepsije povezanih sa lokalizacijom. Karbamazepin i difenin su lekovi izbora; valproati, lamotrigin i barbiturati su rezervisani. Valproati su posebno efikasni u slučaju sekundarnih generaliziranih napadaja. Uz neefikasnost monoterapije primenjuje se polieterapija - kombinacija gore navedenih lekova. Puna otpornost napada na tretman ARD-a je razlog za razmatranje pitanja hirurške intervencije.
Prognoza PE ovisi o prirodi strukturnog oštećenja mozga. Česti napadi otporni na terapiju značajno pogoršavaju socijalnu adaptaciju pacijenata. Napadi koji potiču iz dodatne motorne zone su obično otporni na tradicionalni AED i zahtijevaju hirurški tretman.
Opći principi liječenja epilepsije
Trenutno se razvijaju općeprihvaćeni međunarodni standardi za liječenje epilepsije, koji se moraju promatrati kako bi se poboljšala učinkovitost liječenja i poboljšao kvalitet života pacijenata.
Lečenje epilepsije može se započeti tek nakon što se ustanovi tačna dijagnoza. Termini "pre-epilepsija" i "profilaktički tretman epilepsije" su apsurdni. Prema većini neurologa, liječenje epilepsije treba početi nakon drugog napada. Jedan paroksizam može biti "slučajan", uzrokovan groznicom, pregrijavanjem, intoksikacijom, poremećajima metabolizma i ne odnosi se na epilepsiju. U ovom slučaju, trenutno imenovanje AED-a ne može biti opravdano, budući da su ovi lijekovi potencijalno visoko toksični i ne koriste se u svrhu "prevencije". Prema tome, AED-ovi se mogu koristiti samo u slučaju ponovljenih neprovociranih epileptičkih napadaja (tj. Po definiciji epilepsije).
Dijagnoza epilepsije je uspostavljena i odlučeno je da se imenuje AED. Od 1980-ih, princip monoterapije se čvrsto etablirao u kliničkoj epileptologiji: olakšanje epileptičkih napada treba obaviti prvenstveno jednim lijekom. Trenutno je u potpunosti dokazana nedosljednost starog koncepta imenovanja velikog broja AEP-a istovremeno u malim dozama. Politerapija je opravdana samo u slučaju rezistentnih oblika epilepsije i ne više od 3 AED u isto vrijeme.
Izbor AEP ne treba da bude empirijski. AEP se propisuje strogo u skladu sa oblikom epilepsije i prirodom napada. Uspeh lečenja epilepsije u velikoj meri je određen tačnošću sindromološke dijagnoze (Tabela 1).
AEP se propisuju, počevši od male doze, sa postepenim povećanjem kako bi se postigla terapijska efikasnost ili pojavljivanje prvih znakova nuspojava. U isto vrijeme, klinička efikasnost i podnošljivost lijeka (tab. 2) je odlučujuća.
U slučaju neefikasnosti jednog leka, treba ga postepeno zameniti drugim AEP, efikasnim u ovom obliku epilepsije. Uz neefikasnost jednog AEP-a ne može se odmah dodati drugi lijek, tj. Otići na polieterapiju, ne koristiti sve rezerve monoterapije.
Indikacije za određivanje sadržaja AEP u krvi. Određivanje nivoa AEP u krvi može biti korisno u kliničkoj praksi u sljedećim slučajevima:
Pojava znakova opijenosti;
Nedostatak efikasnosti kada se koriste adekvatne doze;
Prisustvo stanja kod pacijenta koji uzrokuje povredu farmakokinetike AEP-a (bolesti jetre i bubrega, trudnoća, rano doba, itd.);
Preporučuje se u svim slučajevima upotrebe fenitoina (nelinearna farmakokinetika);
Pacijent koristi nekoliko AEP ili drugih lijekova koji mijenjaju farmakokinetiku AEP;
Medicinski pregled.
Treba uvek imati na umu da ne postoji jasna direktna veza između kliničke efikasnosti leka, njegove tolerancije i koncentracije u krvi. U tom smislu, klinički kriterijum efikasnosti i podnošljivosti treba uvijek dominirati laboratorijskim pokazateljima.
Upotreba drugih lijekova pored anti-epileptika u liječenju IE je predmet rasprave. Kod nekih pacijenata koje smo ispitivali u kompleksnoj terapiji korišćeni su vaskularni (cinarizin, cavinton, sermion, tanakan); nootropni (nootropil, pantogam, cogitum, oxybral); metabolički (esencijalno-forte, folna kiselina) lekovi. Njihov uticaj na učestalost napada i opšte stanje pacijenata nije analiziran. Prema apsolutnoj većini stranih istraživača, lekovi ovih grupa nemaju nikakvog uticaja na tok epilepsije i ne koriste se za ovu bolest. Međutim, u nekim slučajevima, kombinacija epilepsije sa glavoboljama, oslabljenim pamćenjem i koncentracijom, toksični efekti AED, upotreba vaskularnih, nootropnih i metaboličkih lijekova mogu biti opravdani. Međutim, ne postoji izgovor za “patološku” upornost nekih lekara koji pacijentima propisuju 5-8 različitih lijekova kao što su diuretici, apsorpcija, vitamin, itd. bez ikakvog zdravog razuma. Trebalo bi strogo raspravljati o imenovanju bilo kojeg lijeka.
Principi otkazivanja tretmana
AEP se može otkazati nakon 2,5-4 godine potpunog nedostatka napada. Klinički kriterijum (nedostatak napada) je glavni kriterij za prekid terapije. U većini idiopatskih oblika epilepsije, povlačenje lijekova može se provesti nakon 2,5 (rolandička epilepsija) - 3 godine remisije. U teškim rezistentnim oblicima (Lennox-Gastautov sindrom, simptomatska parcijalna epilepsija), kao i kod juvenilne mioklonične epilepsije, ovaj period se povećava na 3-4 godine. Sa trajanjem pune terapijske remisije od 4 godine, lečenje treba poništiti u svim slučajevima. Prisutnost patoloških promjena u EEG-u ili pubertetskom periodu bolesnika nisu faktori koji odgađaju ukidanje AEP-a u odsustvu napadaja više od 4 godine.
K.V. Voronkova, doktor medicinskih nauka, profesor Ruskog nacionalnog medicinskog univerziteta. N.I. Pirogov "Ministarstvo zdravlja Ruske Federacije, Moskva Ključne riječi: epilepsija, dijagnoza epilepsije, tipovi napadaja, oblici epilepsije u djece, febrilni napadaji.
Ključne riječi: epilepsija, dijagnoza epilepsije, tipovi napadaja, epilepsija u djece, febrilne konvulzije.
Uvod
Epilepsija je grupa uobičajenih bolesti mozga koje manifestiraju konvulzije. U svijetu više od 60 miliona ljudi pati od epilepsije, općenito u populaciji - otprilike 0,5–1%. U Rusiji - oko 2 miliona djece i odraslih, au SAD - 2,7 miliona pacijenata. Prema nekim studijama, epilepsija je češća kod muškaraca nego kod žena.
Epilepsija je jedna od najčešćih bolesti u djetinjstvu, koja je češća nego, na primjer, bronhijalna astma ili tuberkuloza. U 2/3 svih slučajeva, epilepsija počinje prije 20. godine života. Općenito, epilepsija kod djece se smatra benignim stanjima koja mogu proći sama ili u pozadini terapije u pubertetu, ali to nije slučaj. Mnoge epilepsije u djetinjstvu mogu biti teške za liječenje i praćene su ozbiljnim poremećajima u višoj mentalnoj sferi. Mnoge epilepsije kod djece se ne prepoznaju na vrijeme i ne tretiraju se. Hiperdijagnoza se može primijetiti kada se takva stanja kao što su enureza, poremećaji ponašanja itd. Ekstrapoliraju na epilepsiju, što ima negativne posljedice po dijete.
Također treba napomenuti da je dječja epileptologija izuzetno višestruka, postoje oblici koji se ne primjećuju kod odraslih. Pored toga, kod dece, manifestacija epilepsije je pod uticajem mnogih specifičnih faktora. Kod djece je veliki broj oblika epilepsije istaknut debutom u određenoj dobi (oblici epilepsije ovisni o dobi) - od neonatalnog perioda do adolescencije. Debitno doba je naznačeno u ime ovih oblika epilepsije. Na primjer, benigne konvulzije novorođenčadi, dječja odsutnost-epilepsija, benigna mioklonična epilepsija djetinjstva, itd. Za ove oblike bolesti, početak napada je jedan od važnih znakova koji omogućava ispravnu dijagnozu. Mnoge manifestacije epilepsije mogu se promijeniti s godinama i biti povezane s oštećenjem sazrevanja mozga. Na primjer, infantilni grčevi (Westov sindrom) - napadi u obliku "klimanja", "preklapanja" ("Salaamski napadi"), po pravilu, počinju u prvih 6 mjeseci života i primjećuju se samo u prvih 1-1,5 godina života. Kod dece starije od 1–2 godine, ovi napadi prelaze ili se transformišu u druge: napade padova, napade „bledi“, tonički i toničko-klonički konvulzivni napadi, itd. Kod takvih pacijenata često se primećuje intelektualni poremećaj. S druge strane, tipični absani koji počinju u djetinjstvu ili adolescenciji (kao dio dječje i adolescentske epilepsije) su znak više “benignih” oblika epilepsije, obično ne praćenih smanjenjem inteligencije i drugih poremećaja nervnog sistema. Doba debija i tok ovih oblika epilepsije, uključujući moguću evoluciju, je genetski određen proces.
Ovaj članak bavi se problemom dječije epilepsije u širokom spektru medicinska praksaNe samo neurolozi i psihijatri se mogu suočiti s manifestacijama epilepsije, već i sa liječnicima raznih specijalnosti, a posebno pedijatrima. Nesumnjivo, zadatak pedijatra je samo prepoznati manifestaciju epilepsije među patološkim simptomima i poslati dijete na konsultaciju specijalistu.
Dajte dva klinička primjera poteškoća u diferencijalnoj dijagnozi pedijatrijske epilepsije. Oba primjera također ukazuju na to da je moguća kašnjenja u dijagnostici, koja se iz objektivnih razloga može napraviti s vremenom i pojavom dodatnih „dokaza“. Isto važi ne samo za dijagnozu epilepsije kao takve, već i za uspostavljanje forme. Ponekad je potrebno mnogo godina da se bolest pojavi u vezi sa mogućom transformacijom i evolucijom epilepsije, koja je takođe češća kod dece nego kod odraslih.
Devojka, 12 godina . Sa 8 godina poremećena retka vrtoglavica. Nakon debija vrtoglavice, nekoliko mjeseci kasnije, počele su se pojavljivati paroksizmalna stanja, počevši od vrtoglavice i straha, nakon čega je uslijedila adverzija glave i tijela desno i pad, dok je djevojčica bila svjesna. Trajanje takvih stanja iznosilo je 1-5 minuta, a učestalost je bila jednom tjedno ili mjesec dana. Posmatran od strane neurologa na mjestu boravka nekoliko godina s dijagnozom "vegetativne disfunkcije", propisana je nootropna i vaskularna terapija, ali bez pozitivnog efekta. U dobi od 12 godina, došlo je do porasta paroksizama (do nekoliko puta tjedno), pogotovo kada je nedostajalo sna i uzbuđenja, u zagušljivoj sobi. Takođe, pojavile su se generalizovane toničko-klonične konvulzivne napade sa komponentom versive, nakon čega sledi povraćanje i produženi san nakon spavanja. Neuroimaging je otkrio difuznu zonu promenjenog signala u sivoj materiji desnog parijetalnog režnja. Video EEG monitoring je sproveden na pozadini deprivacije sna, u zagušenoj prostoriji i na pozadini stresa, tokom kojeg je na EEG-u razvijen status neželjenih epileptičkih napadaja, praćenih ictalnim obrascima. Djevojčica je hospitalizirana u bolnici gdje je dijagnosticirana "simptomatska lupinalna epilepsija, statusni tijek" i odabrana antikonvulzivna terapija (valproična kiselina), nakon čega su konvulzije postale sve rjeđe, status epileptičkih napadaja se nije ponovio.
Devojko, 10 godina . Od ranog djetinjstva, neurolog je primijetio sindrom deficita pažnje. Debitacija bolesti zabilježena je sa 9 godina, u snu noću pojavila se tonička napetost lijeve polovice mišića lica uz dodatak kloničnog trzanja mišića udova. Sličan napad ponovio se nakon 2 mjeseca, ali je bio duži (do 10 minuta). Većina napada je povezana sa spavanjem. Djevojka je provela noćni video EEG monitoring, tijekom kojeg su snimane tzv. benigna epileptiformna pražnjenja detinjstva na levoj i desnoj centralno-temporalnoj i zadnjoj plohi. Dijagnostikovana je benigna epilepsija djetinjstva sa centralno-temporalnim adhezijama, rolandskim. Nakon imenovanja antikonvulzivne terapije (valproična kiselina), napadaji se nisu ponovili, koncentracija pažnje se poboljšala, sposobnost učenja, emocionalna labilnost smanjena, djevojčica je postala bolja u školi.
Karakteristična grupa epilepsije
Definicija epilepsije. Epilepsija je hronična bolest mozga koju karakterišu ponovljeni neprovocirani napadi motoričkih, autonomnih, senzornih i mentalnih poremećaja koji nastaju usled prekomernog neuralnog pražnjenja. Ovu definiciju daje glavno telo koje reguliše rad lekara epileptologa - Međunarodna liga protiv epilepsije (Međunarodna liga protiv epilepsije - ILAE), osnovana pre više od 100 godina.
Iz definicije slijedi nekoliko zaključaka:
- Veoma je važan kriterijum za ponovnu pojavu kliničkog događaja, na koji se lekar može osloniti, dijagnostikujući "epilepsiju". Međutim, postoje situacije kada se dijagnoza postavlja nakon prve epizode.
- Epilepsija je spontani (neprovocirani) napadaj. Napadi koji se javljaju samo pod dejstvom faktora koji izazivaju situaciju uslovljavaju situaciju: groznica kod male djece (febrilne konvulzije), poremećaji metabolizma (npr. dijabetes), upotreba alkohola ili droge, itd. Ova kategorija uključuje i konvulzije koje se dešavaju na pozadini bolesti mozga (neuroinfekcije, vaskularne lezije, traumatske povrede mozga). Izuzetak su refleksni oblici epilepsije, koji se odlikuju specifičnim metodama izazivanja napada. To su: fotosenzitivna epilepsija (u kojoj su napadi uzrokovani ritmičkim treperenjem svjetlosti), čitanje epilepsije, epilepsija hrane, izazivanje napada sa taktilnom stimulacijom, razne igre, određeni misaoni procesi itd.
- Pored generaliziranih toničko-kloničkih napadaja (koji se tradicionalno povezuju sa konceptom "epilepsije"), opisani su mnogi različiti tipovi napadaja. Naprimjer, napadi u vidu povrede vizualne percepcije (vizuelne iluzije), kratkotrajno oštećenje pamćenja, osjećaji straha, nasilne misli, napadi u obliku abdominalnog bola, povraćanje, gušenje i drugi koji nisu praćeni konvulzijama. Ako pacijent, posebno dijete, ima takve napade, liječnici često ne mogu prepoznati svoju epileptičnu prirodu dugi niz godina. Često se dijagnoza uspostavlja tek nakon spajanja konvulzivnih napadaja kao rezultat progresije bolesti, što se može vidjeti iz navedenih kliničkih primjera. Kod nekih oblika epilepsije kod jednog pacijenta mogu se pojaviti različiti tipovi napada.
- Osnova svih kliničkih manifestacija epilepsije je uvijek patološki, izrazito snažan iscjedak u živčanim stanicama moždanog korteksa mozga, koji se može registrirati elektrofiziološkim dijagnostičkim metodama - elektroencefalografijom ili EEG-om.
Dijagnoza epilepsije
Dijagnoza "epilepsije" sastoji se od tri komponente (tzv. Kliničko-elektro-anatomski pristup u dijagnostici).
- Kliničke manifestacije napadaja (u nastavku ćemo analizirati napadaje i oblike epilepsije koje se često nalaze u praksi pedijatrijskog neurologa) i istorije.
- Promjene u bioelektričnoj aktivnosti mozga (elektroencefalografija - EEG, video-EEG monitoring). EEG studiju treba obaviti na svim pacijentima sa sumnjom na epilepsiju. Na pacijentovom EEG-u za epilepsiju moguće je identificirati epileptiformne promjene (u obliku kompleksa s valovima šiljaka ili vrha, pojedinačne šiljke, polispike, oštre valove, komplekse valova s usporenim valom, itd.) Koji odgovaraju lokalizaciji i distribuciji. abnormalan iscjedak u korteksu.
Štaviše, najinformativnija i najsavremenija metoda je dugoročno snimanje EEG-a u različitim funkcionalnim stanjima (budnost, san) uz video snimanje onoga što se događa sa pacijentom - video-EEG monitoring. Video EEG monitoring omogućava ne samo da se izvrši preciznija dijagnoza epilepsije, već i da se prate rezultati liječenja.
U ovom odeljku potrebno je razmotriti termin „niska konvulzivna spremnost“, koji se često opisuje encefalogramom, što dovodi do prevelike dijagnoze i postavljanja antikonvulzivne terapije u slučajevima kada dijete nema epilepsiju. Trenutno se ovaj izraz smatra netačnim. Tačniji opis utvrđenih kršenja. U većini slučajeva, lečenje se propisuje samo kada pacijent ima napade, praćenu epileptiformnom aktivnošću na EEG-u. Po pravilu, ako se otkriju promene na EEG-u, a nema kliničkih manifestacija napada, lečenje ne treba propisati. Kada se sprovodi EEG za bilo kakve pritužbe (kao što su glavobolja, itd.), Ali bez kliničkih znakova epilepsije, epileptiformna aktivnost se otkriva u 2-3% slučajeva.
Međutim, postoje uslovi iz takozvane grupe epileptičnih encefalopatija, kada sama aktivnost epileptiforme ima negativan uticaj na kognitivne funkcije djece i ne može se manifestovati na svim epileptičkim napadima. Od posebnog značaja je studija tokom sna, kada epileptiformna pražnjenja zamenjuju fiziološku bioelektričnu aktivnost. Otpuštanja trpe neuronske mreže odgovorne za čuvanje informacija, gradeći program djelovanja. Deca mogu izgubiti svoje stečene veštine, izgubiti govor, postati hiperaktivna, autistična, a nastava sa takvim pacijentima postaje neefikasna. Takvi oblici bolesti otkriveni su posljednjih godina. To su: Landau-Kleffner-ov sindrom (stečena epileptička afazija), ESES (električno stanje usporenog sna), autistična epileptiformna regresija, itd. Takva stanja se nazivaju i kognitivna dezintegracija. U ovim izuzetnim slučajevima, promene u EEG obrascu, čak iu odsustvu napada, služe kao indikacija za razmatranje propisivanja antiepileptičkih lekova. - Promene u strukturi mozga otkrivene metodama neuro-snimanja - kompjuterizovana tomografija (CT), magnetna rezonanca (MR), uključujući MRI visoke rezolucije u režimu epileptičkog skeniranja. Prema svjedočenju primijenjene i druge tehnike.
- U nekim slučajevima, prema indikacijama, vrši se genetičko istraživanje i analiza biohemijskih markera bolesti iz grupe metaboličkih poremećaja (aminoacidopatija, mitohondrijska, peroksizomalna oboljenja, itd.).
Razmotrite manifestacije epilepsije, koje se mogu javiti kod djece - napadaji. Riječ "napad" se ponekad koristi za opisivanje drugih kratkotrajnih poremećaja koji nisu povezani s epilepsijom: nesvjestica, psihogeni napadi, noćni strahovi, izljevi ljutnje itd. Stoga je poželjno koristiti termin "epileptički napad" kada je u pitanju epilepsija. S druge strane, u medicinskom vokabularu postoje mnogi drugi termini koji se koriste i za opisivanje epileptičkih napadaja - epizode iktala, konvulzije, grčevi, grčevi, paroksizmi, epizode "zatvaranja" (svijesti), napada, itd.
Epileptički napad je kratkotrajna epizoda patoloških prekomjernih pražnjenja živčanih stanica moždane kore, uzrokujući stanje koje je primjetno ili neprimjetno za osobu koja ima napad, ili za druge ljude.
Kliničke manifestacije napada su veoma različite kod različitih pacijenata. Epileptički napadaji često imaju iznenadni početak i obično se zaustavljaju spontano, tj. Obično su kratki, traju od nekoliko sekundi do nekoliko minuta i često su praćeni periodom pospanosti ili konfuzije (period nakon napada). Postoje opšteprihvaćene klasifikacije epileptičkih napadaja - Kyoto, 1981, i već su razvijene u XXI vijeku.
Kao prvo, izdvajamo pojmove kao fokalni ili fokalni napad i generalizaciju. Fokalni epileptički napadi su napadi koji počinju sa iste hemisfere. Jednostavni (bez gubitka svijesti) i kompleksni (sa gubitkom svijesti) fokalni napadaji trenutno nisu istaknuti. Ipak, predlaže se procjena oštećenja svijesti u vrijeme napada. Fokalni napadi se mogu transformirati u sekundarne generalizirane. U ovom slučaju, oni govore o auri (aura je, u stvari, početak napada). Aura je veoma važna, jer ukazuje na lokalnu prirodu napada i ukazuje na lokalizaciju epileptičkog fokusa u cerebralnom korteksu. Identifikacija fokalnih napadaja zahtijeva pojašnjenje njihove etiologije (porijekla). Generalizovani epileptički napad - napad koji počinje istovremeno sa dvije hemisfere mozga. Generalizirani napadaji se dijele na konvulzivne i nekonvulzivne i značajno variraju u kliničkim manifestacijama i težini.
Kliničke manifestacije fokalnog napadaja ovise o području korteksa u kojem se odvija pražnjenje. Primjeri uključuju lokalne napade (na primjer, u lice, u ruku), autonomne poremećaje (crvenilo ili bljedilo lica, bol, nedostatak daha, pojačano znojenje, vrtoglavicu, mučninu, čak i mnogo sati izoliranog povraćanja), kao i senzibilizaciju (neobični vizualni, slušni, ukusa, mirisa, taktilnih osjeta, iluzija ili halucinacija) ili mentalnih poremećaja (neobične misli, strahovi, agresija, promjene u stvarnosti onoga što se događa, itd.).
Pacijent sa gubitkom svijesti za vrijeme fokalnog napada može povući odjeću, vrtjeti objekte u rukama, lutati besciljno („automatsko“ ponašanje ili „automatizam“). Smacking usne ili žvakanje, mrmljanje, grimasa, svlačenje i izvođenje besciljnih akcija mogu se posmatrati izolirano ili u različitim kombinacijama. U nekim slučajevima, pojava vanjske agresije i autoagresije. Treba naglasiti da pacijent sve ovo obavlja nevoljno, u stanju promenjene svesti, ne seća se postignuća.
Fokalni napadaji sa sekundarnom generalizacijom (sekundarni generalizirani napadaji) su fokalni napadaji u kojima se epileptički iscjedak širi na korteks u obje hemisfere mozga s razvojem generaliziranog napadaja s pojavom napadaja (češće - tonički-klonički, rjeđe - druge vrste napadaja), gubitkom svijesti , ponekad sa ugrizom jezika, mogućom inkontinencijom urina (a ponekad i izmetom). Ako se pražnjenje širi veoma brzo, onda se spoljašnjim manifestacijama može teško razlikovati sekundarni generalizirani napad od primarne generalizovane. U ovim slučajevima, samo EEG (video-EEG monitoring) će pomoći da se razjasni vrsta napada. Ponekad takozvani simptomi gubitka nakon napada mogu signalizirati doktoru o fokalnoj prirodi napada.
Nova klasifikacija detaljno opisuje nove tipove fokalnih napada.
Na primjer, želatinozni napadaji se češće opisuju s hipotalamičkim hamartomom i manifestiraju se kratkim ili dugim (do statusnog) smijeha u kombinaciji s fokalnim napadima s motoričkim simptomima, mioklonijama, aksijalnim toničnim napadima i fleksorskim grčevima. Konvulzije najčešće debiju prije 5 godina, au slučaju povezanosti s procesima u frontalnim ili temporalnim režnjevima mozga, debitant se bilježi nakon 5 godina. EEG nije specifičan (interiktalni i ictal) - regionalni šiljci, mogu se primijetiti generalizirana pražnjenja.
Generalizirani toničko-klonički napadaji (GTCS) - najčešći tip generaliziranih napadaja. Ranije su ih označavali pojmovi "grand mal" napadaji, ili "veliki konvulzivni napadaji", koji su većini ljudi poznati kao epilepsiju. Pre napada, moguć je period u vidu opšte neugodnosti, slabosti, ponekad traje i satima. Dakle, pacijent može osjetiti da će se napad dogoditi uskoro, ali ne može predvidjeti točno vrijeme njegovog razvoja. Grčevi obično prestaju nakon nekoliko minuta. Nakon završetka napada počinje period nakon napada, ponekad se mogu razviti pospanost, zbunjenost, glavobolja, mučnina i povraćanje. Nakon prestanka napada, često dolazi do dubokog sna, koji se ponekad može pogrešno protumačiti kao nesvjesno stanje. Nakon buđenja, bolesni se ne sjećaju što se dogodilo; pospanost i zajednička bolni bolovi u mišićima, povezana sa prekomjernom mišićnom aktivnošću tokom napada.
Absanse koji su se ranije zvali petit mal (mali napadi). Napadi ovog tipa počinju u djetinjstvu i adolescenciji. Po pravilu, ne nalaze se u drugim starosnim grupama. Pojava izostanaka kod odraslih zahtijeva isključivanje fokalnih napadaja (jer ponekad fokalni napadaji mogu nalikovati odsustvu u njihovim vanjskim manifestacijama). Sa razvojem napada, pacijent iznenada prestaje, izgled izgleda prazan, odsutan (jednostavno odsustvo); Moguće je drhtanje kapaka, gutanje i opuštanje glave ili pad glave na grudi, crvenilo ili bledilo lica, automatski pokreti (kompleksno odsustvo). Napadi su veoma kratki (traju nekoliko sekundi), i pacijent i drugi ih ne primećuju. Odrasli mogu ove epizode interpretirati kao lošu naviku, osobine karaktera, odsutnost djeteta; nazivaju se i „idejama“ (dijete često „misli“), itd. Ponekad se izostanci otkrivaju slučajno kada se dijete ispita o smanjenju školskog uspjeha. To je zbog činjenice da česte izostanke dovode do narušene koncentracije, a zbog čestih “zamračenja”, dijete propušta obrazovni materijal, uprkos tome što je u učionici. Za vrijeme odsutnosti na EEG-u detektiraju se vrlo karakteristične promjene (generalizirana pražnjenja s frekvencijom od 3 Hz ili više). Mogu se izazvati hiperventilacijom (duboko, ritmičko disanje) tokom EEG snimanja. Opisana slika odgovara tipičnoj odsutnosti. Napadi ovog tipa nalaze se u epilepsiji sa dobrom prognozom. Generalno, inteligencija ove djece nije smanjena.
Atipična apsantija se razlikuje manje iznenadno i kraj, svijest je poremećena, ali nije potpuno onesposobljena, dijete izgleda inhibirano. Promene u EEG-u tokom napada se takođe razlikuju od onih sa tipičnim absansom (spora aktivnost pikavasa sa frekvencijom manjom od 3 Hz (obično 2,5–1,5 Hz), difuzna ili generalizovana, ali obično asimetrična, ponekad sa fokalnim početkom, grupna pražnjenja ponekad nepravilan u prirodi sa fluktuacijama svijesti). Atipična apsantija je karakteristična za teške oblike epilepsije sa lošom prognozom, kao što je Lennox-Gastautov sindrom, simptomatska frontalna epilepsija.
Mioklonične napade su vrlo kratke, iznenadne, nevoljne kontrakcije koje mogu zahvatiti cijelo tijelo ili dio njega, kao što su ruke ili glava. Ponekad mioklonični napadi mogu uzrokovati pad pacijenta, ali nakon pada, napad se odmah zaustavlja. Sa miokloničkim napadima koji uključuju ruke, pacijenti iznenada ispuštaju predmete (na primer, pacijent ujutro baca četkicu za zube, ili ujutro pere zube, ili stavlja hranu u doručak). Često se mioklonični napadaji razvijaju ujutro. Pacijenti i rođaci ne idu kod doktora, videći takvo "trzanje", objašnjavajući ih kao manifestacije "neuroze". A ponekad samo pristup generaliziranih toničko-kloničnih napadaja sa padovima vodi pacijenta do lekara, ali na prijemu takvi pacijenti ne mogu prijaviti prethodne napade "trzanja", a doktor u ovom slučaju može pokazati upornost i pita se za dodatne patološke simptome.
Nova klasifikacija naglašava novi tip generaliziranog epileptičkog mioklonusa - masivni bilateralni mioklonus: pojedinačni ili grupirani u nizu aksijalnih vitala cijelog tijela, uglavnom gornjih ekstremiteta, praćeni generaliziranim pražnjenjem poli / šiljaka. Uočava se u adolescentnoj miokloničnoj epilepsiji, sa Draveovim sindromom.
Mioklonija kapaka s apsanima - kontrakcija kapaka, izazvana uglavnom zatvaranjem očiju i fotostimulacijom u simptomatskim i idiopatskim oblicima epilepsije. Pretpostavlja se da mioklonske kontrakcije kapaka sa absansom mogu biti nezavisan, genetski determinisan oblik epilepsije, koji debitira u djetinjstvu sa dugim ili doživotnim kursom.
Stoljeće Myoclonus bez Absanse - javlja se u prvim sekundama pražnjenja EEG-a i uključuje više od tri kontrakcije kapaka, podizanje očiju prema gore, njihovo vertikalno trzanje, trzanje obrva, glava, devijacija glave i očiju. Ponekad postoji mioklonija u rukama.
Epileptički negativni mioklonus - zaustavljanje mišićne aktivnosti (bez prethodne mioklonije) povezano sa vremenom sa šiljkom na EEG u kombinaciji sa fokalnim, toničkim, generalizovanim toničko-kloničnim ili atoničkim napadima, apsanima, grčevima. Posmatrano s rolandičnom epilepsijom, električnim stanjem sporog spavanja, progresivnom mioklonusnom epilepsijom, sa simptomatskom epilepsijom. Može se posmatrati kao komplikacija anti-epileptičke terapije, a onda se ovaj fenomen naziva “pogoršanje epilepsije”. Treba napomenuti da je negativni mioklonus zapravo fokalni inhibitorni napad, a ne generalizovan.
Mioklono-atonički napad (ranije - mioklonsko-astatički) je varijanta mioklonije, kada je drhtanje praćeno gubitkom mišićnog tonusa tokom mioklono-astatske epilepsije. Trzanje je povezano sa generalizovanim pražnjenjem, atonija je povezana sa generalizovanim sporim talasom.
Atonični i tonički napadi su rijetki tipovi napadaja (manje od 1% svih epilepsija). Oni se nalaze u nekim teškim oblicima epilepsije sa početkom u ranom detinjstvu, kao što je Lennox-Gastautov sindrom. Atonički napadi manifestiraju se iznenadnim gubitkom mišićnog tonusa, bolesnik "opušta" i pada (polako "staje"). Konvulzivna smanjenja su odsutna. Tokom toničnog napadaja, tonusa mišića, naprotiv, povećava se, mišići postaju vrlo napeti ("kao kamen"), što je praćeno i padom pacijenta, zbog čega može nastati povreda. Tonični i atonični napadi su često rezultat ozbiljnog oštećenja mozga. Predlaže se da se razlikuju dvije varijante atoničnih napadaja - kratka (sa kratkim gubitkom svijesti ili bez nje, sa naglim porastom nakon atonije) i produžena (gubitak svijesti i generalizirana atonija traju do nekoliko minuta).
Oblici epilepsije u djetinjstvu
Treba napomenuti da se u djetinjstvu mogu pojaviti gotovo svi oblici epilepsije. Međunarodna klasifikacija epilepsijskih i epileptičkih sindroma (1989) je opšte prihvaćena i pristupačna, u kojoj su postavljena dva osnovna principa prema kojima su svi epilepsijski i epileptički sindromi kategorizirani prema etiologiji (idiopatska, simptomatska, kriptogena) i po prirodi (parcijalni (fokalni) i generalizirani). a) napadaji. Pored toga, uzimaju se u obzir debi godine, vodeći tip napada i prognoze. U XXI veku predloženi su novi pristupi klasifikaciji epilepsije - ovo je katalog poznatih, starosnih klasifikacija, itd.
Nećemo se baviti fokalnim simptomatskim oblicima epilepsije, koji su najčešći kod odraslih bolesnika s epilepsijom, ali često i kod djece. Semiologija napadaja u ovim oblicima je dobro poznata i može biti u korelaciji sa lokalizacijom patološkog procesa i širenjem procesa i epileptiformne aktivnosti na druge dijelove mozga.
Konvulzivni napadi koji se javljaju kod male djece (od 6 mjeseci do 6 godina) u pozadini visoka temperaturanazivaju se febrilni napadi. Takvi napadi se ne smatraju epilepsijom i odnose se na situaciono izazvane napade. To je zbog činjenice da se napadi u ovom slučaju dešavaju samo u određenoj situaciji (porast temperature) i samo u određenoj starosti. Spontani napadi (koji nisu povezani s tjelesnom temperaturom) u tim slučajevima ne bi smjeli biti. Pregled (uključujući pregled neurologa, procena razvoja deteta i EEG izvan napada) ne otkriva patološke promene. Dugotrajna antiepileptička terapija nije propisana za ovu djecu, iako postoji velika vjerovatnoća ponovnog pojavljivanja febrilnih napadaja u kasnijim bolestima djeteta, praćenih groznicom. Preporučuje se izbjegavanje podizanja temperature, primjena metoda smanjenja temperature lijekova i neliječenih u pozadini zaraznih bolesti; u nekim slučajevima, lekovi za lečenje epilepsije se propisuju profilaktički za period akutnih zaraznih bolesti. Sa godinama (nakon 5–6 godina) febrilne napade nestaju bez traga. Međutim, u nekim slučajevima, febrilne napade su prva manifestacija epilepsije i dalje su praćene pojavom spontanih epileptičkih napadaja koji nisu povezani sa groznicom. Granicu između febrilnih konvulzija koje ne zahtijevaju liječenje i epilepsije, koje zahtijevaju dugotrajnu antiepileptičku terapiju, određuje liječnik.
Epileptička encefalopatija ranog djeteta sa supresivno-eksplozivnim promjenama na EEG - Otahara sindromu (CO). U CO, prenatalno oštećenje mozga je uočeno u većini slučajeva. Bolest počinje u dobi od 3 mjeseca, češće - u 1. mjesecu života djeteta. Glavni tip napadaja su tonički grčevi u trajanju od 10 sekundi. Često se pojavljuju kao serija (10-40 grčeva se može pojaviti u jednoj seriji). Ukupan broj grčeva može dostići 300-400 dnevno. Takođe su primećeni kratki fokalni napadaji. Uočeni su zakašnjeli mentalni i motorički razvoj, poremećaji u neurološkom statusu. Odlikuje se otpornošću na terapiju i lošom prognozom bolesti sa visokim mortalitetom u ranom djetinjstvu. Moguća je transformacija u Westov sindrom.
Westov sindrom (SV) . Nova klasifikacija odnosi se na skupinu epileptičnih encefalopatija u dobi od 20 godina. SV karakteriziraju sljedeći kriteriji: poseban tip epileptičkih napadaja - infantilni grčevi, promjene na elektroencefalogramu u obliku gipsaritmije, odgođeni psihomotorni razvoj. Sa godinama, većina slučajeva SV se transformiše u fokalne oblike epilepsije, rjeđe u Lennox-Gastaut sindrom. Otkrivene su promene: odlaganje mentalnog i motoričkog razvoja.
Lennox-Gasto sindrom (SLH) . Ovaj oblik epilepsije prema novoj klasifikaciji spada u skupinu epileptičnih encefalopatija djetinjstva. Bolest debituje u uzrastu od 2 do 8 godina sa vrhuncem od 3-5 godina. U oko 20% slučajeva SLH se transformiše iz Westovog sindroma. Febrilni napadaji prethode razvoju sindroma u 10% slučajeva. U SLG-u su uočeni različiti napadi: tonični aksijalni, miatonske kapi, atipični absans, epileptički status “malih motoričkih napadaja”, mioklonska, generalizirana konvulzivna, fokalna. Status epilepticusa javlja se kod 75% bolesnika sa SLH. Često mogu postojati mentalni poremećaji.
Benigna epilepsija u detinjstvu sa centralnim temporalnim adhezijama - rolandska epilepsija . Ovaj oblik epilepsije počinje u dobi od 3–14 godina (u 85% slučajeva, u dobi od 4-10 godina). Preovladavaju faringo-oralni paroksizmi: grlo zvuči kao "grgljanje", hipersalivacija (povećana salivacija), anarthria (poremećaj govora) sa intaktnom sviješću. U isto vrijeme je moguće osjećati "bockanje" na nebu, sluznicu usta; trzanje usana ili jezika. Često se javljaju fokalni motorički klonički napadaji sa lokalizacijom u mišićima lica, kao iu ruci, rjeđe uz uključivanje nogu na istoj strani i sekundarno generalizirane. Napadi su rijetki i kratki. Karakterizira se svojim izgledom prilikom buđenja ili zaspanja. Intelekt ne pati. Neka djeca imaju smanjenu koncentraciju s hiperaktivnošću. Na EEG-u su identifikovani specifični znaci - benigni epileptiformni ispadi iz detinjstva (DERD) sa lokalizacijom u centralnim temporalnim tragovima. Karakteristično je povećanje epileptiformne aktivnosti tokom perioda sporog sna. Prognoza je često povoljna, ali se može javiti i atipična evolucija epileptičkog statusa uspavanog sna (opisana gore).
Benigna epilepsija u detinjstvu . Postoje dva različita sindroma: sa ranim (3-6 godina) debi, Panayotopoulos varijantom i kasnim (obično 6-14 godina) početkom, Gasto varijantom. Bolest se manifestuje izolovanom vizuelnom aurom, autonomnim-visceralnim simptomima (mogu biti mučnina, povraćanje i drugi simptomi), fokalni motorički napadi. Karakterizira se pojava napada nakon zaspanja i prije buđenja. Tu je glavobolja, povraćanje (ovi simptomi su takođe mogući na kraju napada), adverzija (skretanje) očiju i glava u stranu; hemiklonske konvulzije (konvulzije jedne polovice tijela); moguće sekundarne generalizacije. Panayotopulos sindrom se manifestuje napadima sa produženim gubitkom svesti. Starija djeca doživljavaju vizualne senzacije - elementarne vizualne halucinacije u izolaciji ili prije početka „velikog napada“ ili prelaska na druge vrste napada. Učestalost napada je niska.
Landau-Kleffner-ov sindrom (SLC), ili "sindrom stečene epileptične afazije", prema novoj klasifikaciji, spada u grupu epileptičnih encefalopatija detinjstva. SLK počinje u dobi od 3–7 godina. Do početka bolesti motorički, mentalni i govorni razvoj pacijenata je prikladan uzrast. Poremećaji govora se postepeno razvijaju, počevši od poremećaja govora, zatim se pridružuje motorna afazija. U slučaju SLC-a mogu se primijetiti epileptički napadaji: fokalni motorički napadi, atipične odsutnosti, atopični, mioklonični i sekundarni generalizirani konvulzivni paroksizmi. Kod 25% pacijenata, SLK se javlja u odsustvu epileptičkih napada ili prisustvu pojedinaca u anamnezi. Fokalni simptomi ne postoje u neurološkom statusu. Kod EEG-a, epileptiformni poremećaji se otkrivaju u 100% slučajeva. Morfologija epiaktivnosti odgovara DER-u. Epileptiformna aktivnost se povećava u snu. Prognoza za SLK u vezi sa epileptičkim napadajima: kod skoro 100% pacijenata napadaji su potpuno zaustavljeni i epileptiformna aktivnost je blokirana početkom puberteta (pod uticajem AEP ili spontano). A prognoza o poremećajima govora, kognitivnih funkcija i ponašanja nije tako povoljna.
Dečji apsces epilepsija (DAE) . Debi odsustva u epilepsiji u dječjem apscesu javlja se u dobi od 3 do 9 godina. Kod devojčica je ovaj oblik epilepsije češći. Komplicirane odsutnosti su više karakteristične za AED, posebno sa pokretima glave (lagano spuštanje leđa i trzanje), pomicanjem oka prema gore, u trajanju od 3 do 30 sekundi (obično 5-15 sekundi) i učestalošću od nekoliko desetina i stotina dnevno. Pre kraja napada, pacijenti mogu imati kratke automatizme (udaranje, premlaćivanje itd.). Hiperventilacija (duboko i učestalo disanje) je glavni faktor za pojavu izostanaka. Generalizirani konvulzivni napadaji su uočeni u približno 25% bolesnika s AED.
Juvenilna apscesna epilepsija (UAE) . Debi odsustva u epilepsiji adolescentnog apscesa - obično u 9-13 godina. Apsorpcija u SAE je obično kraća u trajanju i rjeđe u frekvenciji nego u AED. Karakteristično je prevlast jednostavnih izostanaka, tj. Bez motorne komponente, koja traje oko 6 sekundi. U 65–90% slučajeva, pacijenti sa UAE imaju generalizirane konvulzivne napade.
Epilepsija sa izolovanim generaliziranim konvulzivnim napadima . Debitacija bolesti je uočena u širokom rasponu godina. Klinički se ispoljava samo kod jednog tipa napadaja - generaliziranih konvulzivnih napadaja.
Juvenilna mioklonična epilepsija (UME, Janzov sindrom) . Debitovanje juvenilne mioklonične epilepsije primećeno je u uzrastu od 11-15 godina. Djevojke češće pate. Glavni simptom su mioklonični napadi, koji su lokalizovani uglavnom u ramenom pojasu i rukama, uglavnom u mišićnim grupama ekstenzora, a rjeđe hvata mišiće nogu. Spašena je svijest tijekom miokloničkih napada. Karakteristično je pojavljivanje ili povećanje napadaja u prvim minutama i satima nakon buđenja pacijenata. U 90% slučajeva, beleži se i AHEC, au 30% - odsustvo. Čimbenici koji izazivaju napade u UME su deprivacija sna, naglo nasilno buđenje, ritmičko treperenje svjetlosti itd.
U zaključku Treba napomenuti da je dječja epilepsija vrlo raznolika. Potrebno je obratiti pažnju i na paroksizmalne simptome i na različite vrste poremećaja u višoj mentalnoj sferi. Ako postoji sumnja na epilepsiju, dijete treba uputiti na konsultaciju i pregled kod specijaliste.
Reference: